
Abstract
Rationale
Oxidative stress is a major contributor to brain damage in patients with ischaemic stroke. Uric acid (UA) is a potent endogenous antioxidant molecule. In experimental ischaemia in rats, the exogenous administration of uric acid is neuroprotective and enhances the effect of rtPA. Moreover, in acute stroke patients receiving rtPA within 3 h of stroke onset, the intravenous administration of uric acid is safe, prevents an early decline in uric acid levels and reduces an early increase in oxidative stress markers and in active matrix metalloproteinase nine levels.
Aim
To determine whether the combined treatment with uric acid and rtPA is superior to rtPA alone in terms of clinical efficacy in acute ischaemic stroke patients treated within the first 4.5 h of onset of symptoms.
Study design
Multicentre, interventional, randomised, double-blind and vehicle-controlled efficacy study with parallel assignment (1:1). Estimated enrolment: 420 patients over 3 years, starting in January 2010. Treatment arms included patients will receive a single intravenous infusion of 1 g of UA dissolved in a vehicle (500 ml of 0.1% lithium carbonate and 5% mannitol) (n = 210) or vehicle alone (n = 210). Inclusion and exclusion criteria: the study will include patients older than 18 years, treated with rtPA within the first 4.5 h of clinical onset and with a baseline National Institute of Health Stroke Scale score >6 and <25 and a modified Rankin Scale score ≤ 2 before the ischaemic event. Patients with renal insufficiency, gout or asymptomatic hiperuricaemia treated with allopurinol will be excluded.
Study outcomes
The primary outcome measure is the proportion of patients achieving an modified Rankin Scale of 0 to 1 at 3 months after treatment or two in those patients with a prior qualifying modified Rankin Scale of 2. Secondary outcome measures include the final infarction volume measured at 72 h and the proportion of patients with symptomatic intracranial haemorrhage (≥ 4 points of increase in the National Institute of Health Stroke Scale score).
Introduction and rationale
Currently, rtPA is the only approved therapy for stroke patients within the first hours of clinical onset (1). Oxidative stress is a major contributor to brain damage in patients with ischaemic stroke, particularly when ischaemia is followed by reperfusion (2). Uric acid (UA) is an endogenous product derived from the metabolism of purines, which, in humans, is responsible for 60% of the total antioxidant capacity of the organism (3). Previously, we had reported an observational study indicating that higher levels of UA at stroke admission predicted a better outcome at follow-up (4). Recent experimental evidences have shown that the exogenous administration of UA is neuroprotective both in cortical and in subcortical brain areas as a result of its antioxidant properties. In these studies, animals treated with UA showed smaller brain infarction after transient focal ischaemia, both using the intraluminal model and after the injection of autologous clots (5 6). Moreover, in the thromboembolic model, the protective effects of the exogenous infusion of UA were additive to the effects of rtPA (alteplase) (6). Likewise, a recently published pilot trial showed that the administration of UA was free from serious adverse effects in stroke patients receiving rtPA within 3 h of stroke onset (7). In that trial, the infusion of UA (1 g) avoided an early decay in UA levels and reduced lipid peroxidation markers (malondialdehyde) and the activation of matrix metalloproteinase-9, a molecule related to poor clinical outcomes (7 8). Based on these data, we aim to conduct a phase 3, randomised, double-blind and controlled trial assessing the clinical efficacy of UA administration in acute ischaemic stroke patients treated with rtPA within the 4.5-h window.
Methods
Design
The URIC-ICTUS trial is a multicentre, interventional, randomised, double-blind, vehicle-controlled and efficacy study with a parallel assignment (1 : 1) including acute stroke patients receiving intravenous (i.v.) rtPA within the first 4.5 h of the onset of symptoms. The study will be conducted according to ICH/GCP guidelines. All participating institutions have obtained Local Ethics Committee approval before trial initiation. The URIC-ICTUS Study is registered at www.clinicaltrials.gov/ (registration number NCT00860366).
Patient population - inclusion and exclusion criteria
Patients fulfilling all the following criteria will be included in the study:
age older than 18 years acute ischaemic stroke treated with rtPA within the first 4.5 h of clinical onset baseline National Institute of Health Stroke Scale (NIHSS) <6 and <25, and modified Rankin Scale (mRS) ≤ 2 before the stroke absence of blood in the CNS in the initial cranial CT signed informed consent.
Otherwise, patients will be excluded if they disclose one or more of the following exclusion criteria:
presence of any of the current exclusion criteria for the administration of rtPA in the clinical practice a history of gout with or without a history of gouty nephropathy, or uric lithiasis asymptomatic hyperuricaemia under chronic treatment with allopurinol or chronic treatment with lithium chronic renal insufficiency (baseline creatinine <1.5 mg/dl or 132.6 (μmol/l).
Randomisation
A block randomisation list will be generated by means of the PROC PLAN module of SAS (version 9). Randomisation will be stratified by centre and baseline NIHSS (NIHSS score of 10 as a cut point) with a probability of assignment to each treatment arm of 1 : 1. The randomisation will be performed in a web-based system provided by Onmedic Networks S. L. (Barcelona, Spain), under the supervision of the Clinical Trials Unit of Hospital Clínic of Barcelona.
Treatments
Patients will be randomised into two treatment arms. Patients allocated to the active arm will receive an i.v. infusion of 1 g of UA dissolved in a vehicle containing 500 ml of 0.1% lithium carbonate and 5% mannitol over 90 min. Patients allocated to the control arm will receive a single intravenous infusion of 500 ml of a vehicle containing 0.1% lithium carbonate and 5% mannitol over 90 min.
The selected dose of UA is based on previous clinical studies including healthy volunteers (9 10), and acute stroke patients receiving rtPA (7). The intravenous infusion of doses up to 1 g of UA has been shown to be safe and to reduce several parameters of oxidative stress. In acute stroke patients receiving rtPA, UA infusion in the early phase prevents an early decay in UA levels and reduces markers of lipid peroxidation and the activation of matrix metalloproteinase-9, a molecule related to poor clinical outcomes (7 8).
Uric acid will be obtained from Sigma (Ultra pure preparations; Sigma Chemical Co., Poole, UK). Both UA and vehicle solutions will be manufactured by Laboratorios Grifols (Barcelona, Spain) following Good Manufacturing Practices regulations. The use of UA as an investigational medicinal product in human clinical trials received recently the approval of the Spanish Medicines and Healthcare Products Agency (eudraCT 2004-002937-37).
A schematic outline of the study including the timing of treatments is shown in Fig. 1. Intravenous rtPA treatment will be initiated at a standard dose and regimen (0.9 mg/kg, initial bolus of 10% of the final dose and the remaining dose as an intravenous infusion lasting 60 min) following current clinical guidelines. When a patient meets all the inclusion/exclusion criteria and signs the informed consent, UA/vehicle infusion will be initiated as soon as possible after starting the infusion of rtPA treatment. The 90-min intravenous infusion of the experimental treatment (UA/vehicle) will be performed through a peripheral venous line in an extremity different from that used for rtPA infusion. Antithrombotic treatments will be avoided during the first 24 h after rtPA treatment, except for special circumstances (e.g. pulmonary embolism).
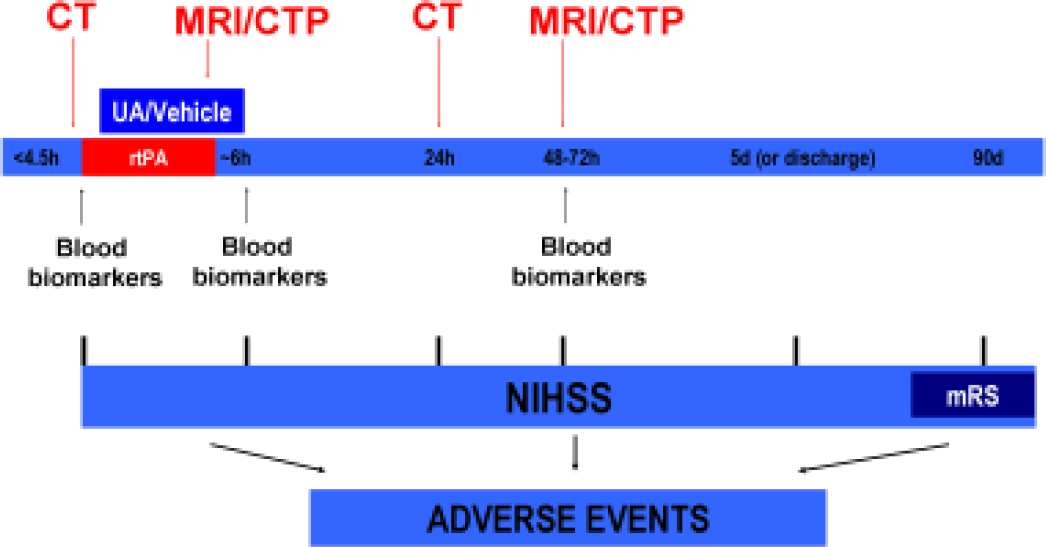
URICO-ICTUS study design. Schematic outline of the study including the timing of treatments and ancillary studies.
Primary outcome
The primary outcome measure of the study is the proportion of patients with a favourable clinical outcome at 3 months, defined as achieving an mRS score of 0–1 at 3 months after treatment, or 2 in those patients showing an mRS of 2 before the inclusion in the study. The evaluators of the functional outcome at day 90 will be certified in the use of neurological scales (NIHSS, mRS) and will use a structured interview questionnaire to homogenise the evaluation in all participating centres.
Secondary outcomes
Secondary outcome measures include the following efficacy and safety variables:
proportion of patients with NIHSS <2 at 2 h after finishing the experimental treatment proportion of patients with NIHSS <1 at day 90 proportion of patients achieving a Barthel scale of 95 to 100 at day 90 all-cause mortality within the first 90 days final infarction volume measured by means of MRI or multimodal CT at 72 h of onset (in selected centres) proportion of patients with an intracranial haemorrhage associated with a worsening of at least 4 points in the NIHSS within the first 36 h of treatment.
Ancillary studies
Neuroimaging substudy
All patients will receive a baseline CT scan before the inclusion in the study to discard the presence of any intracranial haemorrhage and to evaluate the extension of early signs of ischaemia (ASPECT score) and the presence of the hyperdense middle cerebral artery sign. In selected centres, a multimodal CT (angio-CT and CT-perfusion) or MRI (including DWI, PWI, T2, T1, *T2 and FLAIR sequences and angio-MRI) will be performed at the end of rtPA infusion (40–120 min after the start of the infusion) and at 72 h. The variables that will be evaluated in the multimodal imaging include arterial status (early vs. late recanalisation and TICI score), the presence and volume of mismatch, the volume of established infarction and the presence of intracranial haemorrhages. An additional mandatory CT scan will be performed 24 h after rtPA infusion to evaluate the presence of intracranial bleeding. The neuroimaging studies will be evaluated in a single centre by neuroradiologists blinded to treatments and to the clinical course of patients.
Blood biomarkers’ substudy
Following the recommendations of the Stroke Academy Industry Roundtable (STAIR) (11), a longitudinal blood biomarker study will be executed to evaluate the biological activity of the investigational drug. In selected centres, serial blood extractions will be performed at baseline, at 6–12 h, and at 48 h after the start of the experimental treatment (Fig. 1). After appropriate processing of the blood, serum and plasma samples will be stored at −80°C until their analysis, which will include biochemical markers of parenchymal damage (S100B), markers of blood–brain barrier damage (matrix metalloproteinase-9, fibronectin) and oxidative stress markers (malondialdehyde, F2-isoprostanes).
Data-safety monitoring board (DSMB)
The study will be monitored by the Clinical Trials Unit of Hospital Clinic of Barcelona. A DSMB will be composed by three independent members (one neurologist, one biostatistician and one clinical pharmacologist), who will evaluate periodically the frequency and type of fatal or serious adverse events, including the rate of death of any cause, the incidence of symptomatic intracerebral haemorrhages (increase in at least 4 points in the NIHSS) and all the adverse events considered to be related to the investigational drug.
Sample size
The primary outcome of this trial is a favourable clinical outcome at 90 days defined as an mRS score of 0–1 at 90 day or an mRS of 2 in patients with a qualifying mRS of 2 at entry in the study. Based on an expected response rate of 40% in the primary outcome variable in the control group and assuming a loss to follow-up of 5%, a total of 210 patients per group will be required to detect a significant difference not inferior to 14% between treatment groups (α 5%, β 80%). These estimations are based on the recently published results of the ECASS3 trial (12), on the results of a pilot trial of UA infusion in acute ischaemic stroke patients receiving rtPA (n = 24) (7) and on data from the prospective registry of patients treated with rtPA in the co-ordinating centre (Hospital Clínic of Barcelona; n = 120 at the time of sample size calculation). Patients will be included over a period of 3 years and the analysis of results is planned at the end of 2012.
Statistical analyses
The primary outcome variable will be analysed in the ‘intention-to-treat’ population using a binary logistic regression model adjusted by centre and baseline NIHSS. Additionally, an exploratory unadjusted analysis will also be performed. As a proof of sensitivity and consistency of the results, the ‘per protocol’ analysis will be provided as well. Mortality will be evaluated using a Cox regression model and adjusting by the same variables used in the analysis of the primary outcome variable. The rest of the secondary outcome variables will be analysed using the appropriate hypothesis test (exact Fisher's test for categorical variables, Student's t-test for continuous variables and the Mann–Whitney U-test for ordinal variables). The statistical analysis will performed using spss software version 16.0 (SPSS Inc., Chicago, IL), and significance will be established at the P <0.05 level (two sided).
Study organisation and funding
The URICO-ICTUS trial is funded by the Institute of Health Carlos III of the Spanish Ministry of Health (grant number EC07/90276) and by a private grant from Fundación Doctor Melchor Colet. A total of six Spanish tertiary hospitals will participate in the study. The study will be co-ordinated in the Hospital Clinic of Barcelona by the Fundació Clínic per a la Recerca Biomèdica and the Clinic Trials Unit of Hospital Clínic de Barcelona. The Executive Committee (seven members, principal investigator Ángel Chamorro) is responsible for the development of the study protocol and its amendments. The independent DSMB (three members) is responsible for reviewing safety and efficacy data on an ongoing basis. It will also perform interim analyses (group sequential procedure) on the primary end-point at scheduled time-points during the study according to a prespecified plan.
Summary and conclusions
Growing experimental and clinical evidence prove UA as a promising neuroprotective agent in brain ischaemia. In experimental ischaemia, the benefits yielded by the exogenous infusion of UA are in addition to those provided by rtPA. In both healthy volunteers and patients suffering an acute ischaemic stroke, the i.v. infusion of UA is feasible and safe. In stroke patients treated with intravenous rtPA, the addition of UA is able to reduce the parameters of oxidative stress and prevent the activation of matrix metalloproteinase-9. Based on this knowledge, the URICO-ICTUS trial will evaluate the clinical efficacy of the intravenous infusion of UA in acute ischaemic stroke patients receiving rtPA within the first 4.5 h. Moreover, the preplanned neuroimaging and blood biomarker substudies will quantify signs of the biological activity of UA infusion using several validated surrogate outcomes.