
Abstract
Rationale
Preclinical work demonstrates that the transcription factor peroxisome proliferator-activated receptor gamma plays an important role in augmenting phagocytosis while modulating oxidative stress and inflammation. We propose that targeted stimulation of phagocytosis to promote efficient removal of the hematoma without harming surrounding brain cells may be a therapeutic option for intracerebral hemorrhage.
Aims
The primary objective is to assess the safety of the peroxisome proliferator-activated receptor gamma agonist, pioglitazone, in increasing doses for three-days followed by a maintenance dose, when administered to patients with spontaneous intracerebral hemorrhage within 24 h of symptom onset compared with standard care. We will determine the maximum tolerated dose of pioglitazone.
Study Design
This is a prospective, randomized, blinded, placebo-controlled, dose-escalation safety trial in which patients with spontaneous intracerebral hemorrhage are randomly allocated to placebo or treatment. The Continual Reassessment Method for dose finding is used to determine the maximum tolerated dose of pioglitazone. Hematoma and edema resolution is evaluated with serial magnetic resonance imaging (MRI) at specified time points. Functional outcome will be evaluated at three- and six-months.
Outcomes
The primary safety outcome is mortality at discharge. Secondary safety outcomes include mortality at three-months and six-months, symptomatic cerebral edema, clinically significant congestive heart failure, edema, hypoglycemia, anemia, and hepatotoxicity. Radiographic outcomes will explore the time frame for resolution of 25%, 50%, and 75% of the hematoma. Clinical outcomes are measured by the National Institutes of Health Stroke Scale (NIHSS), the Barthel Index, modified Rankin Scale, Stroke Impact Scale-16, and EuroQol at three- and six-months.
Introduction
Intracerebral hemorrhage (ICH) accounts for 10–15% of all strokes. ICH remains a devastating disease and current treatment options lag far behind those for ischemic stroke. To date, there are no approved therapies that improve outcome and treatment remains mainly supportive. As such, functional outcomes in ICH remain dismal with only 20% of victims being independent at six-months (1).
ICH causes both primary and secondary injury. The primary insult is due to disruption of adjacent tissue with deposition of blood and subsequent mass effect (2). Secondary injury occurs with development of edema, free radical formation, inflammation, and direct cytotoxicity of products deposited during hemorrhage. The effects of persistent blood degradation products and edema on functional recovery are complex and not well-known. Removal of the hematoma prior to red blood cell (RBC) lysis and release of toxic material may limit the deleterious effect on otherwise salvageable brain parenchyma. It appears intuitive that quicker resolution of the hematoma and edema would be associated with more rapid and enduring functional recovery.
One way to reduce the production of blood degradation products is surgical hematoma evacuation. This has been the impetus for trials of hematoma evacuation, which, unfortunately, have not demonstrated benefit (3,4). Other treatment efforts have focused on halting the growth of the hematoma as this factor has been shown to correlate with clinical outcome (5,6). Few studies have focused on the natural history of hematoma absorption and edema resolution (7–10). With current clinical efforts focused on the hematoma in the acute setting, a treatment that could enhance hematoma resolution, thus limiting secondary injury, would be a natural adjunctive treatment to acute therapy for ICH.
Another way to decrease the hematoma burden is to take advantage of the body's own mechanism of hematoma removal, phagocytosis. In our preclinical work, we have demonstrated that the transcription factor, peroxisome proliferator-activated receptor gamma agonist (PPAR-γ), in microglia/macrophages plays an important role in augmenting phagocytosis, while, simultaneously, downregulating the expression of pro-inflammatory mediators and upregulating antioxidative enzymes. We propose that this dual role of PPAR-γ can be used to design a therapeutic approach to treat ICH by promoting hematoma absorption and reducing deleterious pro-inflammatory and pro-oxidative responses, known contributors to secondary brain damage. Clinically relevant PPAR-γ agonists include the thiazolidinediones (TZDs). Pioglitazone (PIO) and rosiglitazone (ROSI) are TZDs currently approved for the treatment of adults with type ii diabetes mellitus.
PPAR-γ has been shown to play a role in regulation of central nervous system inflammation in many models of neurologic disease including experimental autoimmune encephalitis (11), Alzheimer's disease (12), Parkinson's disease (PD) (13), amyotrophic lateral sclerosis (14), ischemic stroke (15–18), and most recently, ICH (19,20). Consistent findings in each of these animal models include downregulation of pro-inflammatory responses, a reduction in the number of reactive microglia and macrophages, and preservation of the integrity of affected cell types (13,1421).
In animal models of ischemic stroke, TZDs decrease infarct volume and are associated with improved neurologic outcomes (15,1722). These findings are consistent across the different classes of PPAR-γ agonists (i.e. troglitazone, ROSI, PIO, and prostaglandin 15d-PGJ2). In addition, the effect on infarct volume is dose-dependent, and above a particular threshold, no additional histological benefit is observed (22,23). The dose-dependent threshold for PIO has not been clearly established as doses of 20 mg/kg in rodent models of ischemic stroke and PD have demonstrated benefit (13,24).
Preclinical work with the autologous injection ICH model in our lab demonstrates that PPAR-γ agonists promote hematoma resolution, decrease neuronal damage, and improve functional recovery (25). We have also shown that PPAR-γ agonists demonstrate the ability to protect other brain cells from the secondary injury induced in a mouse model of ICH. Thus, we propose that PPAR-γ in microglia/macrophages acts as an important factor in promoting hematoma absorption and protecting other brain cells from ICH-induced secondary damage and represents a promising therapeutic target in the management of ICH.
This protocol describes the rationale and design of a dose-escalation and dose-finding study designed to test the hypothesis that escalating doses of PIO at doses that are effective in animal models of stroke can be safely administered to patients in the acute setting of ICH. In addition, we will explore whether the speed of hematoma/edema resolution in ICH represents a radiographic biological marker of activity that can be correlated with clinical outcome and treatment effect of PIO. The ultimate purpose is to provide baseline data on an aspect of ICH that has not been previously targeted for treatment in an effort to develop a safe and effective treatment strategy that may be practical and applicable for both specialized stroke centers and community hospitals.
Methods
Design
Safety of Pioglitazone for Hematoma Resolution in Intracerebral Hemorrhage (SHRINC) is a phase 2 randomized, blinded, placebo-controlled study designed to assess the safety of the PPAR-γ agonist, PIO, in increasing doses from 0·1 to 2 mg/kg/days for three-days followed by a lower maintenance dose, when administered to patients with spontaneous ICH within 24 h of symptom onset compared with standard care. Using the Continual Reassessment Method (CRM) design for dose escalation, we will determine the maximum tolerated dose (MTD) of PIO in patients with spontaneous ICH. The SHRINC study is registered at http://www.clinicaltrials.gov/ (registration number NCT00827892).
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria are listed in Table 1. Initially, the inclusion criteria included patients with a Glasgow Coma Scale (GCS) ≥ 8; however, due to lower than expected enrollment, the protocol was amended to include patients with a GCS ≥ 6.
Study inclusion and exclusion criteria
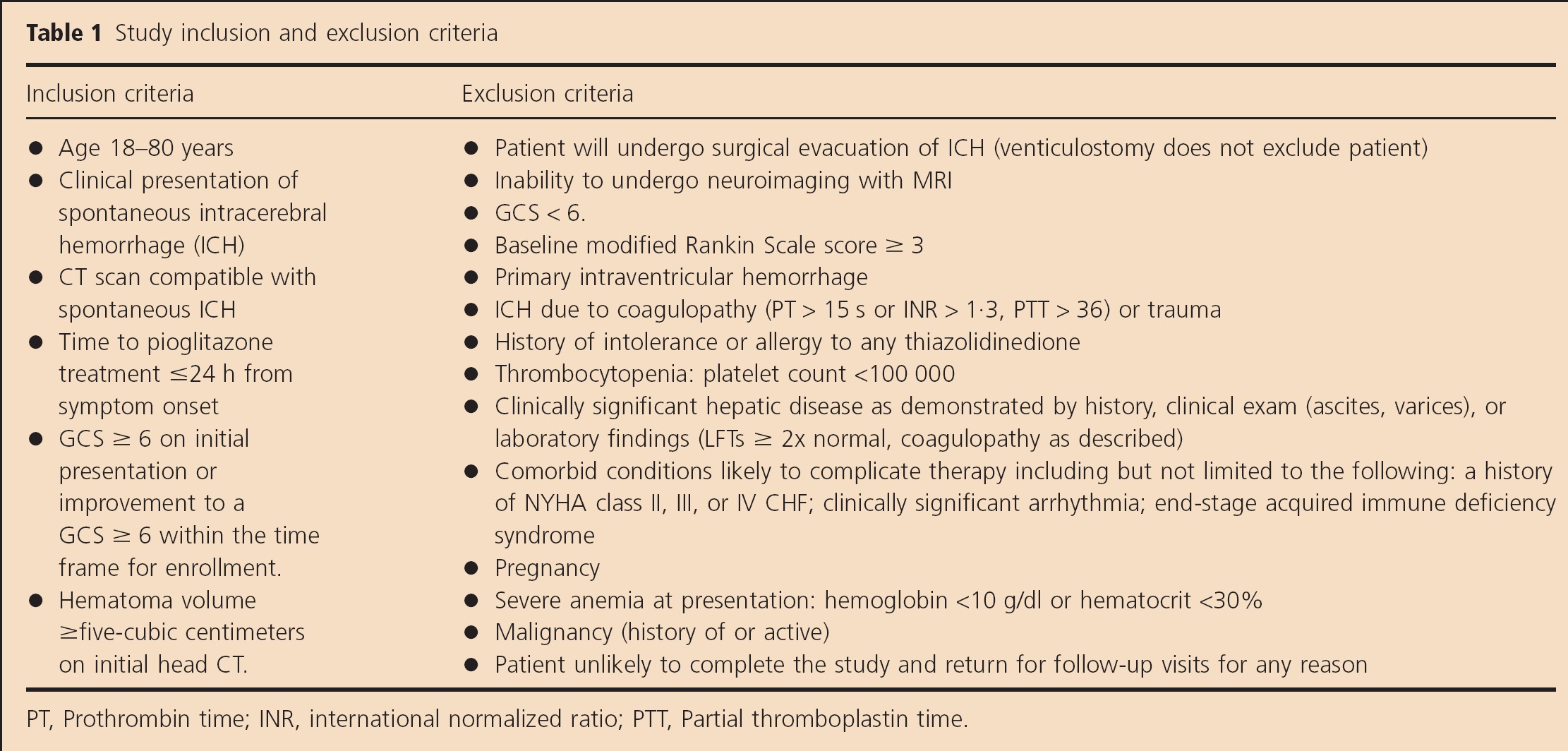
PT, Prothrombin time; INR, international normalized ratio; PTT, Partial thromboplastin time.
The inclusion of a control group is not typical of the usual CRM dose escalation trial design. In this translational pilot study, we have chosen to include a control group for the following reasons: First, many of the adverse events (AEs) and serious AEs (SAEs) we expect to see in the PIO treated group also occur in the ICH population as part of the natural history of disease and comorbidities. The true incidence of these AE/SAEs in our ICH population is difficult to determine based on available information. Thus, we will collect this information prospectively to obtain a more accurate assessment of safety. Second, in our preclinical work, more rapid hematoma resolution was correlated with improved neurologic recovery. This finding has not been demonstrated in the clinical setting. With the use of MRI, we will obtain baseline information regarding the natural history of hematoma resolution and its relationship with clinical recovery. Furthermore, serial MRI in a treated and placebo group will allow us to explore whether treatment changes the natural history of hematoma resolution as demonstrated in our preclinical work. Lastly, traditional CRM studies fix the intercept and estimate the slope of the dose-response curve using a single prior estimate of the beginning dosage with no additional estimates of the natural history of patients not receiving treatment. A by-product of this study's design is the natural history of ICH patients, which provides the additional capability to reestimate the intercept with each cohort of patients, thus obtaining an enhanced approximation of the dose-response relationship.
Randomization
Patients are randomly assigned to either control or treatment in a 1:1 allocation ratio in cohorts of six (three treatment, three placebo). We attempted to avoid an imbalance between the treatment groups due to age, hematoma volume, and milder disease in transferred patients by using an adaptive randomization/minimization schedule that ensures balance between the two study arms with respect to age (<70 years, ≥70 years), hematoma volume (<30 cc, ≥30 cc), and whether the patient presented directly to our emergency department or was transferred from another hospital for each treatment arm based on the proportion of allocated treatments at the time of randomization.
Treatment
Rationale for Choice of Pioglitazone, dose, Timing, and Duration
We have chosen to work with PIO for three reasons. First, PIO is neuroprotective based on data from rodent ischemic models (19,2223) and our ICH rodent model (25) (Fig. 1). Second, the pharmacokinetic properties of PIO allow daily administration, which is important for compliance in the outpatient setting: long half-life of three- to seven-hours for PIO and 16–24 h for its metabolically active metabolites; it has been shown to cross the blood–brain barrier (26). Third, while the neuroprotective effects of the TZDs have been shown to be a class effect, the side-effect profile differs. For instance, troglitazone is associated with an increased incidence of liver toxicity while ROSI and PIO are not. ROSI has been reported to be associated with an increased risk of cardiac toxicity (27,28). PIO, however, has been shown to have a more favorable cardiovascular safety profile in the prospective PROspective pioglitAzone Clinical Trial In macroVascular Events (PROACTIVE) trial (29), a randomized trial of cardiovascular outcomes. Thus, current information regarding safety profile favors the use of PIO in this high-risk patient population.
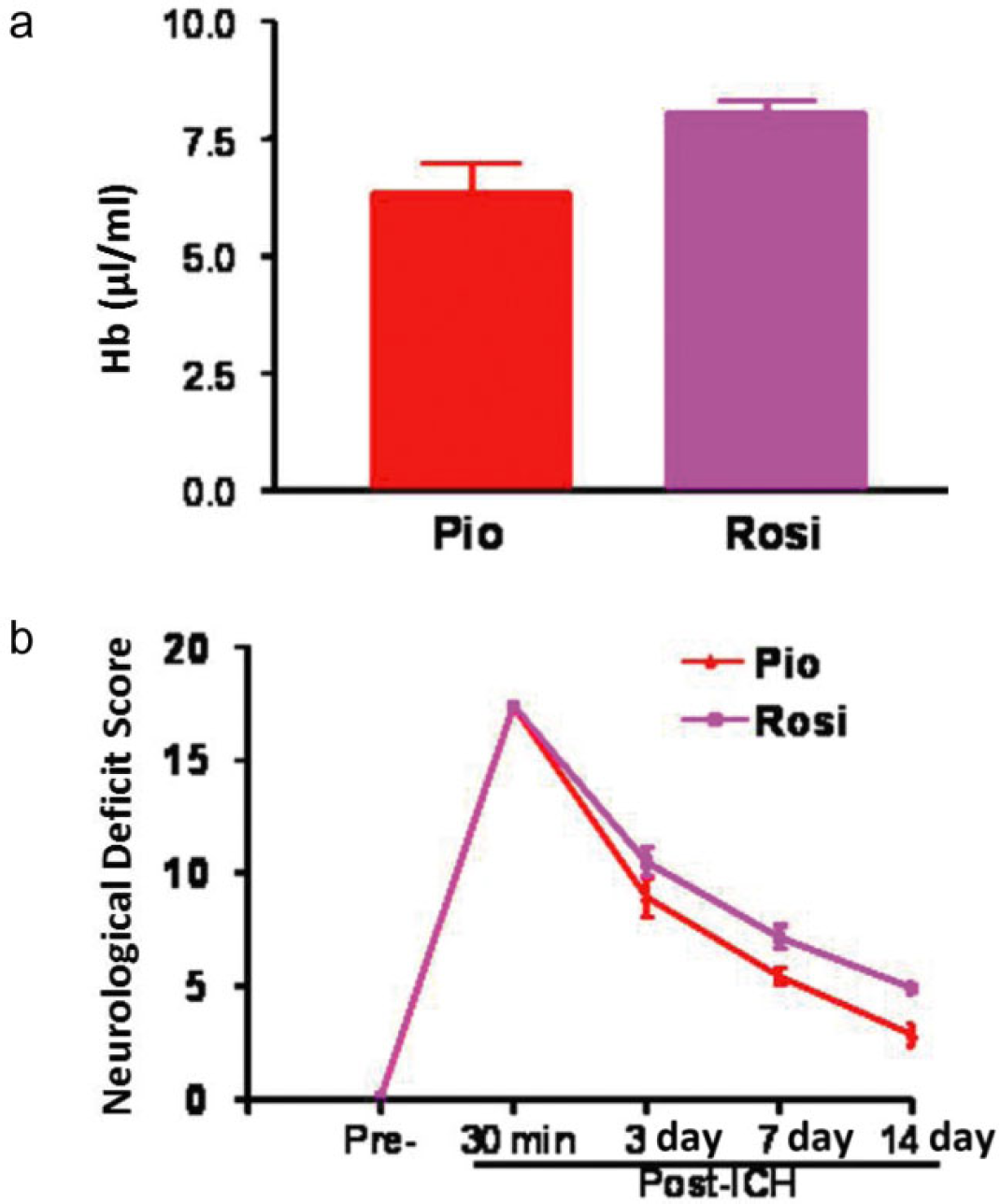
Pioglitazone (PIO) vs. rosiglitazone (ROSI). Comparison of PIO (1 mg/kg, intraperitoneally, n = 7) to ROSI (0·1 mg/kg, n = 7) demonstrates that A. PIO showed a trend toward being more effective than ROSI in improving hematoma absorption, and B. functional recovery at 14 days after ICH. Doses were selected based on our in vitro experiment results (dose with potential to induce PPAR-γ activation) and the dosage regimen used clinically for diabetes patients.
We chose the doses of PIO based on animal model data regarding neuroprotection as well as safety results in humans. The daily recommended dose for diabetic patients is 15–45 mg daily. For a 70-kg person, this translates to 0·2–0·6 mg/kg/day. Preclinical data in both ischemic stroke and ICH demonstrate neuroprotection at doses ranging from one- to four-milligrams per kilogram (22,2330). The package insert for PIO reports that over 8500 patients have been treated with PIO in randomized controlled trials. However, our patient population is acutely ill in contrast to the outpatient population in which safety studies were performed. Lastly, there is limited evidence regarding safety in humans at doses higher than 60 mg daily, which our trial will address in the ICH population.
The ideal timing of PIO therapy for neuroprotection is difficult to determine with certainty from available data. In animal models of ischemic stroke, neuroprotection with TZD therapy was more robust when administered as pretreatment (23) before the stroke, and the benefit may decrease as time to treatment increases. However, in our mouse model of ICH, PPAR-γ agonist administration at 24 h after ICH was equally as neuroprotective as the pretreated group (25). Because this study is focused on safety and dose finding rather than efficacy, a time window of 24 h after symptom onset is used. In ICH, the persistence of toxic RBC degradation products suggests that longer term treatment may be beneficial. In our laboratory studies, microglia are activated as early as one-hour after the ICH (31). In addition, an increase in the number of hematogenous phagocytes occurred over two- to three-days and lasted for at least two-weeks. Thus, we will begin treatment with high-dose PIO as early as possible after the ICH up to 24 h after symptom onset. We plan to use a high-dose of PIO for the first three-days and then decrease the PIO to 30 mg daily for the duration of treatment. In this way, we hope to optimize the benefit of treatment in the acute setting, minimize toxicity while the hematoma is resolving, and decrease the potential of unmasking treatment ‘allocation due to the development of the well-known side effect of PIO, fluid retention’.
Duration of treatment is also difficult to determine with certainty; however, in our preclinical work, we have demonstrated that the hematoma volume is about 108%, 67%, and 32% at three-days, seven-days, and 14 days after ICH, respectively, compared with hematoma volume at 24 h (25). This time frame for resolution was enough to identify both a difference in hematoma resolution and clinical outcome in the treatment group. Thus, with serial MRI, we will explore the time to resolution of 75% of the hematoma, which will provide individualized treatment duration since the actual hematoma size will vary between patients in contrast to the preclinical situation where the hematoma size is uniform.
The intervention in this trial is oral PIO or placebo. Patients allocated to treatment receive escalating doses of encapsulated PIO for three-days followed by a daily maintenance dose of 30 mg. Patients allocated to placebo receive encapsulated lactose for the duration of treatment. A midrange maintenance dose of 30 mg for the treatment group was chosen to minimize the possibility of unmasking treatment allocation. Patients are typically titrated to an outpatient dose of 45 mg PIO over a period of time. Consultants felt that initiation of PIO 45 mg would be more likely than a lower dose to elicit the common side effect of fluid retention and thus unmask treatment allocation. Treatment is discontinued once 75% of the hematoma has resolved (determined with serial MRI) or after 10 weeks of treatment, whichever comes first. Study procedures are outlined in Fig. 2.
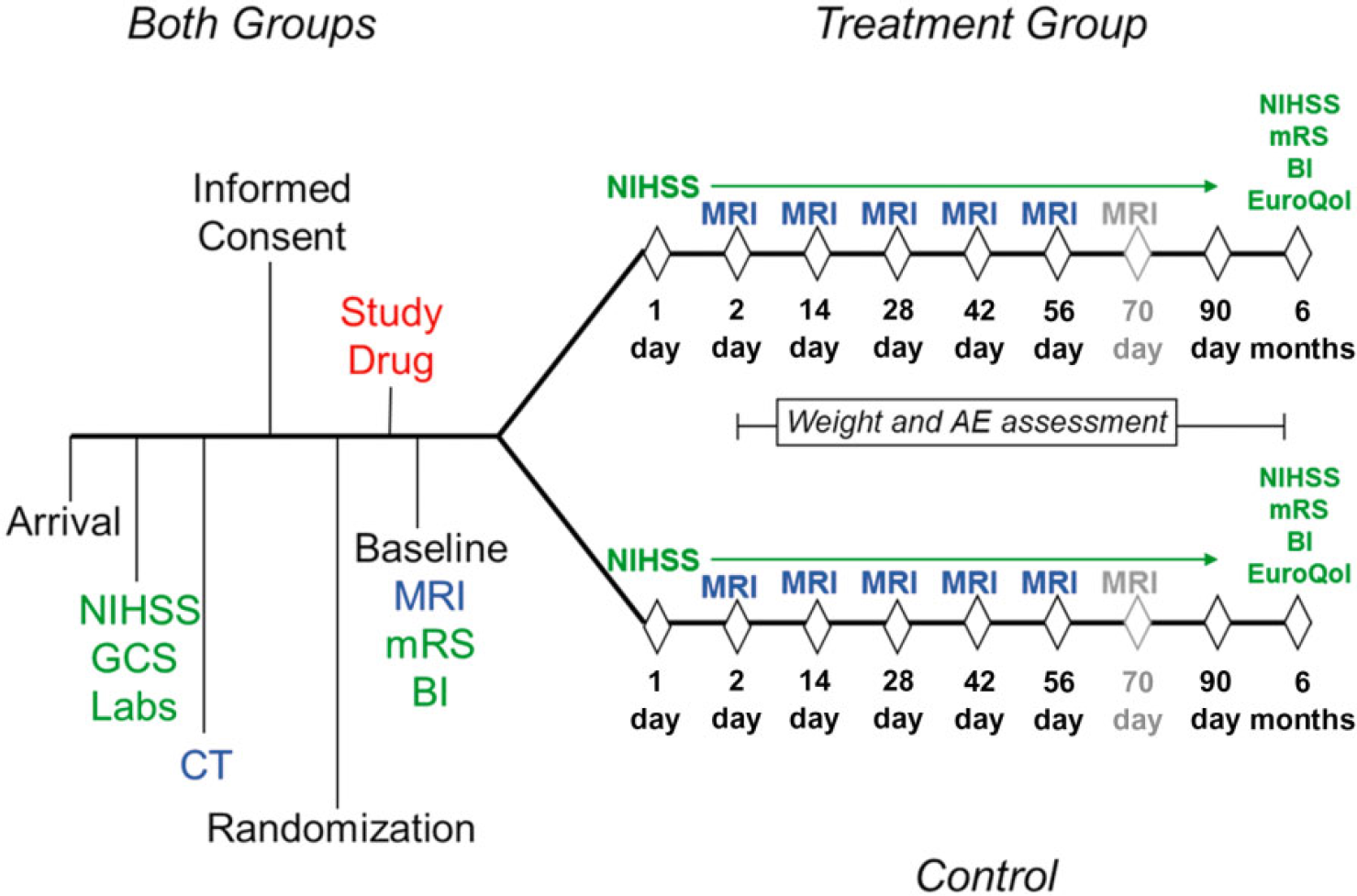
Overview of SHRINC study procedures. At each MRI visit, clinical assessment and AE evaluation will also occur. The addition of neuroimaging at day 70 is reserved to follow edema if still present and for those patients with hematomas that may not resolve as rapidly.
Primary Safety Outcome Measure
The primary outcome measure is mortality rate at discharge or day 14, whichever comes first, of ≤15%. Retrospective review of our registry demonstrates a 15% in-hospital mortality rate for patients with an initial GCS≥8. In determining the primary measure of safety, an analogy with IV recombinant tissue plasminogen activator (rt-PA) for ischemic stroke was considered: the SAE of symptomatic hemorrhage is higher with IV rt-PA with the benefit of improved clinical outcomes and no increase in mortality. Similarly, in this group of patients with ICH, we would be willing to tolerate some level of adverse effects as long as mortality was not increased. Thus, we would not be willing to tolerate a mortality rate any higher than expected with our standard of care. Dose escalation will proceed to the next cohort if discharge mortality is ≤15%.
In this study, a dose escalation/de-escalation decision is made using the CRM calculations (32,33). The first three patients randomized to treatment received dose level 1 (Table 2). After each cohort, the statistician estimates the MTD based on the in-hospital mortality rate for that cohort. There is a potential delay of two-weeks between cohorts in patient accrual since complete information on the enrolled patients is required before enrollment of the next cohort.
Proposed dose escalation
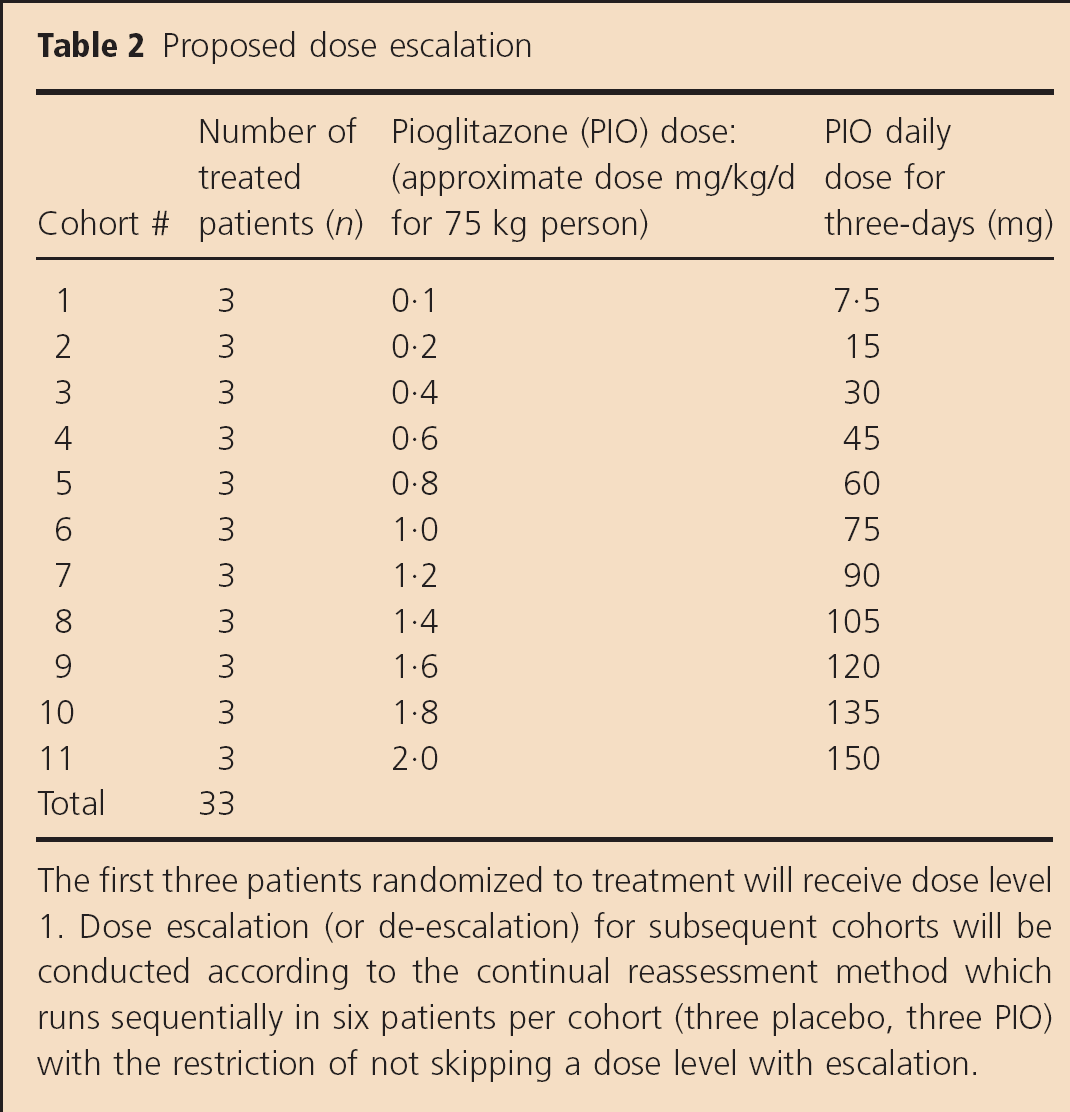
The first three patients randomized to treatment will receive dose level 1. Dose escalation (or de-escalation) for subsequent cohorts will be conducted according to the continual reassessment method which runs sequentially in six patients per cohort (three placebo, three PIO) with the restriction of not skipping a dose level with escalation.
Secondary Outcome Measures
Secondary safety outcome measures include three- and six-month mortality as well as the occurrence of clinically significant AEs/SAEs collected and assessed by personnel blinded to treatment assignment. With the most common side effect of PIO being fluid retention, anticipated SAEs in the acute setting include:
symptomatic cerebral edema: neurologic deterioration defined as an increase in NIHSS of ≥four AND increase in surrounding edema on Computed tomography (CT)/MRI determined by clinician/radiologist
respiratory toxicity: increase in the work of breathing necessitating continuous positive airway pressure (CPAP), Bilevel Positive Airway Pressure (BiPAP), or endotracheal intubation
cardiovascular toxicity: hypotension requiring the use of vasopressors, Congestive Heart Failure (CHF), acute myocardial infarction
hypoglycemia: glucose <50 mg/dl requiring the use of D50; symptoms of hypoglycemia, which are alleviated with glucose administration
hematologic toxicity: anemia or a significant drop in the hemoglobin or hematocrit, which requires the use of RBC transfusion or volume expanders and is not explained by another cause
hepatic toxicity: increase in Liver Function Tests (LFTs) > three times the upper limit of normal
Other secondary outcome measures include the following:
characterization of the speed of hematoma resolution in order to determine duration of treatment
exploration of whether speed of hematoma and edema resolution in ICH represents a biological marker of activity, which can be correlated with radiographic and clinical treatment effect of PIO
estimation of the potential clinical efficacy of PIO. Functional outcome is evaluated at three- and six-months with the NIHSS score, Modified Rankin Scale, Barthel Index, Stroke Impact Scale-16, and EuroQol
Clinical, Laboratory, and Imaging Investigations
The schedule of events is shown in Table 3. Clinical follow-up evaluations are performed by investigators blinded to treatment assignment. Additional clinical assessment and AE/SAE determination occurs at each MRI visit. All patients undergo a brain MRI at baseline, day two, day 14, and day 28. Additional MRIs are planned if 75% hematoma resolution has not occurred at days 42, 56, and 70. Once the patient has reached 75% hematoma resolution or 10 weeks of treatment (whichever comes first), study medication is discontinued. Image data acquisition for SHRINC is composed of a localizer sequence, sagittal T1, axial diffusion weighted imaging with Apparent Diffusion Coefficient (ADC) reconstruction, axial T2 Dual Gradient Recalled Echo, axial Fluid Attenuated Inversion Recovery, axial T2, and axial 3D Circle of Willis Time-of-Flight Magnetic Resonance Angiography with the baseline study to evaluate for vascular abnormality.
Schedule of events
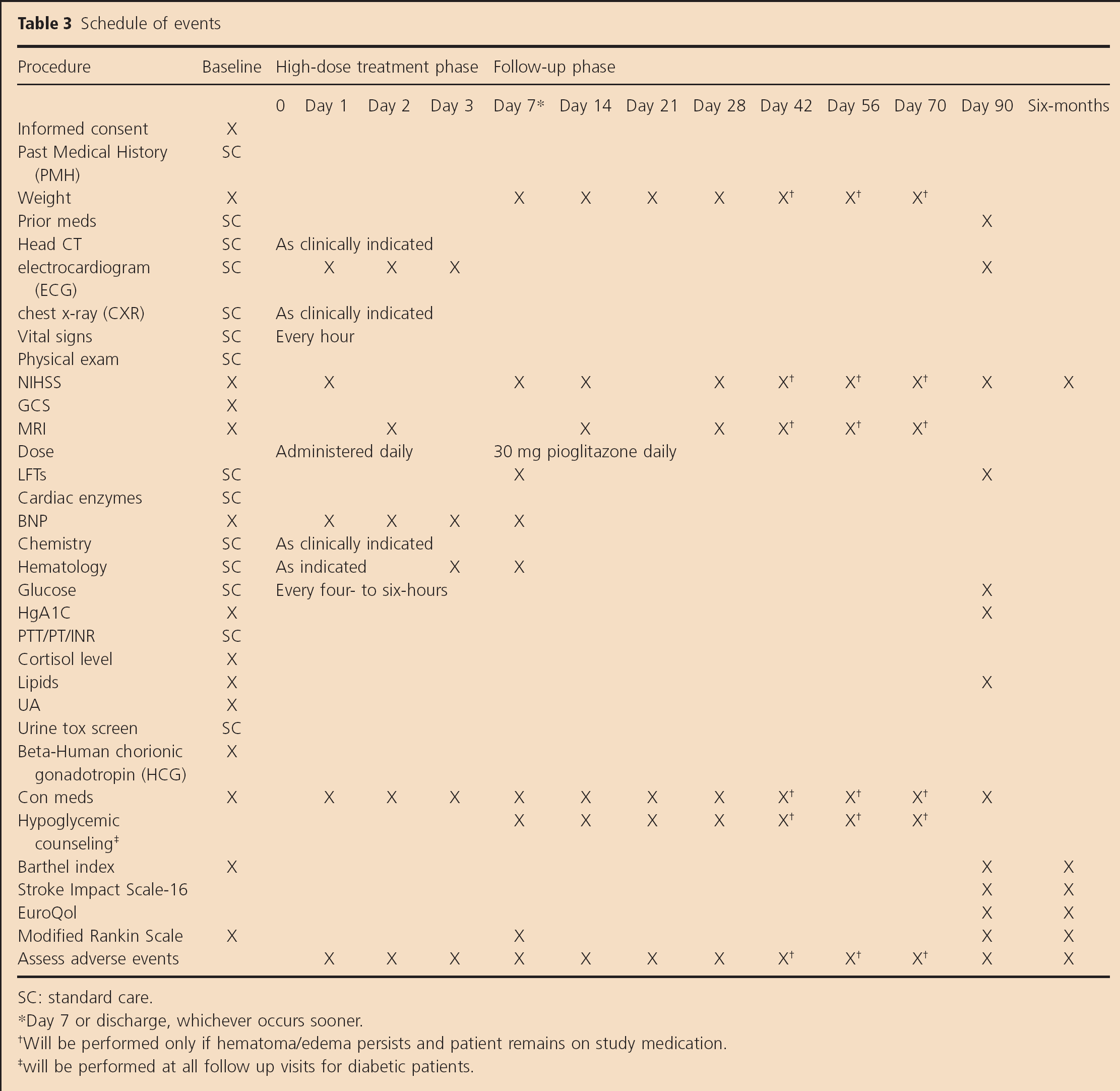
SC: standard care.
Day 7 or discharge, whichever occurs sooner.
Will be performed only if hematoma/edema persists and patient remains on study medication.
will be performed at all follow up visits for diabetic patients.
Data Safety Monitoring Board (DSMB)
Our DSMB is composed of three independent members (one neurologist, one cardiologist, and one biostatistician) who evaluate, periodically, the safety measures in this study as well as accrual rate. After two cohorts were enrolled (12 patients, six in each arm), safety was assessed with the statistical stopping rule utilizing the conditional power approach shown in Table 4. This stopping rule utilizes an SAE rate of 15% with 80 patients enrolled and 70% power. The SAE rate is used to alert the DSMB in the scenario where the study may need to be halted. For example, if the DSMB meets when 30% of the 80 patients have been studied, we would need to have 11 in-hospital deaths in order to be sure that there is a 70% chance that at study completion we would conclude that the in-hospital death rate is >15%. If the DSMB meets when 75% of the study is complete, we would need 16 deaths out of 60 patients to draw the same conclusion. The DSMB is notified if the stopping rule is violated, in which case the trial will be halted pending DSMB review. At that time, the DSMB will determine if the deaths are attributable to PIO prior to proceeding in the trial.
Statistical stopping rule
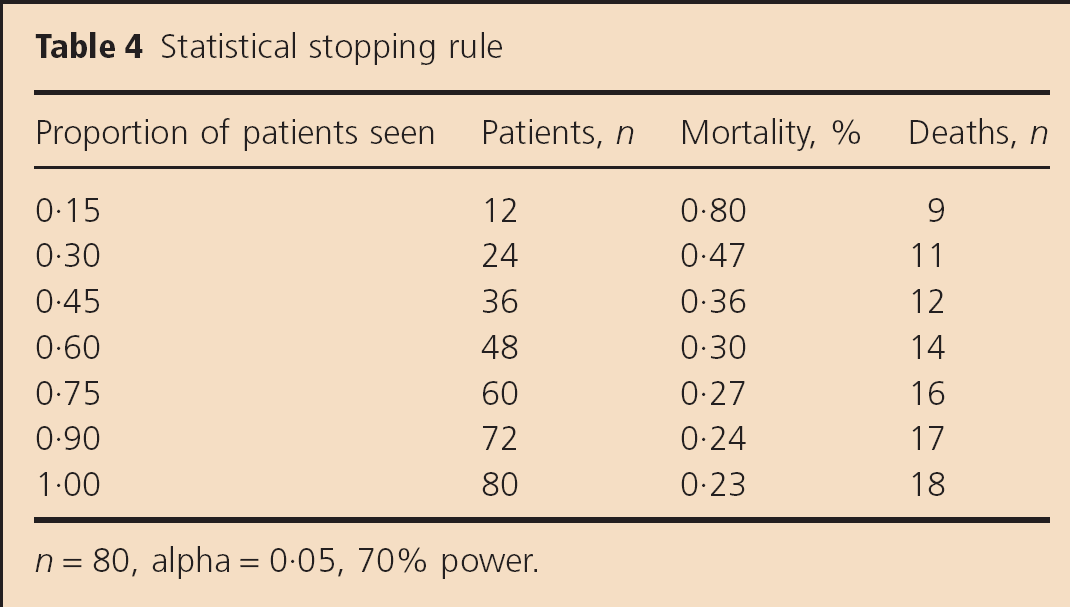
n = 80, alpha = 0·05, 70% power.
Statistical Analysis and Sample Size Calculations
The sample size estimates are a function of the dose-escalation, dose-finding design of the study. Cohorts of three per dose escalation were chosen because this cohort size consistently produces less toxicity since it forces more experimentation at lower dose levels (33); a conservative approach since PPAR-γ agonists have not been investigated in this high risk population. We have planned for up to 11 dose escalations within this design. A sample size of 33 patients in each of the treatment and control groups (total n = 66) allows for 11 dose escalations. Anticipating a 20% dropout rate, we plan to enroll a total of 80 patients (40 treated, 40 control).
A prespecified interim analysis will occur after half of the subjects have been enrolled. The main goal of this interim analysis is to assess whether increasing doses of PIO result in an increase in the likelihood of developing secondary AE/SAEs. In addition, we will evaluate the performance of the randomization program and evaluate whether there is an appreciable relationship between dose and clinical outcomes. Our study is not powered for efficacy; however, these data will provide information that is not currently available in the literature regarding hematoma and edema resolution and will assist in calculating sample size estimates for treatment effect in an efficacy trial.
The analysis plan for the primary outcomes will proceed according to the intention-to-treat principle. If there is significant evidence of imbalance between the treatment and the control arms, we will also conduct analysis that controls for potential confounding variables. Descriptive analysis will be conducted for each of the primary and secondary outcomes and the distributional assumption will be checked thoroughly before adopting the modeling approaches. For repeated measures, the analysis will include generalized estimating equation or random effects modeling. For modeling count data, before applying the Poisson regression model, overdispersion issues will be tested. If overdispersion exists in the observed data, an alternative approach for count data will be explored (e.g. negative binomial regression model). In case of excessive zero counts (e.g. a significant number of patients with no severe AEs), we will adopt a modeling approach that is designed for excessive zeros, e.g. zero-inflated Poisson regression model or zero-inflated negative binomial regression model. In our modeling approach, in addition to including all the significant main effects, the analysis will also control for interaction effect if one exists.
Study Organization, Funding, and Acknowledgments
The SHRINC Trial is funded by Specialized Programs of Translational Research in Acute Stroke (SPOTRIAS) P50-NS044227 from the National Institutes of Health to the University of Texas-Houston Medical School (UT Health) Stroke Program, American Heart Association Clinical Research Program Award 0885050N, and National Institutes of Health (NIH)/National Center for Research Resources (NCRR) 1KL2 RR024149 to the University of Texas-Houston Center for Clinical and Translational Sciences. This is a single-center study at UT Health and Memorial Hermann Hospital in the Texas Medical Center. The authors would like to acknowledge Mitch S.V. Elkind, MD, and Ken Cheung, PhD (Columbia SPOTRIAS Center), for providing assistance during the development phase of the trial, UT Health Center for Clinical and Translational Science for assistance in the development phase of the trial and our DSMB Members: Claudia Pedroza, PhD, David Chiu, MD, Ali E. Denktas, MD, and Robert G. Hart, MD.
Summary
This protocol represents our translational efforts for a novel therapeutic approach to the treatment of ICH: namely, targeted stimulation of phagocytosis to promote efficient removal of the hematoma without harming surrounding brain cells. In addition, we have utilized two adaptive features in our clinical trial:
adaptive randomization to minimize confounding in this small group of patients and
the CRM to efficiently determine a MTD which will inform a future efficacy study.
Despite the clear relationship of hematoma volume with outcome (5,6), hematoma resolution is an aspect of the disease that has not been targeted for treatment before now and data is lacking regarding the natural history of this process and its role in recovery. Hematoma and edema resolution are measurable radiographic findings with MRI technology and represent an opportunity to evaluate radiographic change as a potential biological marker of activity which, if present, may serve as a marker for therapeutic effect in future treatment trials targeting the secondary injury caused by ICH. The multiple mechanisms of action of the PPAR-γ make it a promising therapeutic option for a disease which currently has no approved therapies to improve outcome.