
Abstract
An important proportion of transient ischemic attack or ischemic stroke is attributable to moderate or severe (50–99%) atherosclerotic carotid stenosis or occlusion. Platelet biomarkers have the potential to improve our understanding of the pathogenesis of vascular events in this patient population. A detailed systematic review was performed to collate all available data on ex vivo platelet activation and platelet function/reactivity in patients with carotid stenosis. Two hundred thirteen potentially relevant articles were initially identified; 26 manuscripts met criteria for inclusion in this systematic review. There was no consistent evidence of clinically informative data from urinary or soluble blood markers of platelet activation in patients with symptomatic moderate or severe carotid stenosis who might be considered suitable for carotid intervention. Data from flow cytometry studies revealed evidence of excessive platelet activation in patients in the early, sub-acute, or late phases after transient ischemic attack or stroke in association with moderate or severe carotid stenosis and in asymptomatic moderate or severe carotid stenosis compared with controls. Furthermore, pilot data suggest that platelet activation may be increased in recently symptomatic than in asymptomatic severe carotid stenosis. Excessive platelet activation and platelet hyperreactivity may play a role in the pathogenesis of first or subsequent transient ischemic attack or stroke in patients with moderate or severe carotid stenosis. Larger longitudinal studies assessing platelet activation status with flow cytometry and platelet function/reactivity in symptomatic vs. asymptomatic carotid stenosis are warranted to improve our understanding of the mechanisms responsible for transient ischemic attack or stroke.
Background
Approximately 25–30% of all transient ischemic attacks (TIAs) or ischemic strokes may be attributed to thromboembolism from or less commonly from hemodynamic ischemia distal to a moderate or severe (50–99%) atherosclerotic carotid stenosis or occlusion (1,–3).
Prior studies have indicated that the two-year risk of stroke in the arterial territory supplied by a ≥60% internal carotid artery (ICA) stenosis is approximately 4% in ‘asymptomatic’ medically treated patients in whom the stenosis is incidentally picked up (4,5). More recent data have suggested that the two-year risk of stroke may have fallen to approximately 0.68% in patients with asymptomatic ≥50% ICA stenosis receiving modern secondary preventive treatment (6). However, additional information from transcranial Doppler (TCD) ultrasound monitoring may aid risk stratification and treatment decisions regarding carotid intervention in patients with asymptomatic carotid stenosis (7,8). The recently published Asymptomatic Carotid Emboli Study indicated that the two-year risk of ipsilateral stroke in patients with asymptomatic ≥70% carotid stenosis who have microembolic signals (MESs) detected on TCD ultrasound was 3.6% (7). In contrast, the two-year risk of ipsilateral stroke may be as high as 26% in medically treated patients with ‘symptomatic’ severe (≥70%) carotid stenosis who have had a recent TIA or stroke (9). The recent ‘Clopidogrel plus aspirin vs. aspirin alone for reducing embolisation in patients with acute symptomatic cerebral or carotid artery stenosis’ study revealed that therapy with clopidogrel and aspirin is more effective than aspirin alone in reducing MES in patients with predominantly intracranial symptomatic stenosis (10). Recent studies have illustrated the potential role of MES detection and carotid plaque imaging in risk-stratifying asymptomatic carotid stenosis patients to identify those who may derive most benefit from best medical rather than surgical therapy (11,–13). However, the potential usefulness of adding specific platelet activation and/or reactivity data to these investigations to guide treatment decisions in patients with carotid stenosis is uncertain.
The mechanisms responsible for this disparity in stroke risk between asymptomatic and symptomatic severe carotid stenosis patients are not fully understood but may relate to differences in the morphology of the atherosclerotic plaque, differences in endothelial activation, or differences in the thrombogenicity of the circulating blood (14). Histological studies indicate that the carotid plaque is composed of a dense ‘cap’ composed of connective tissue embedded with smooth muscle cells, with varying amounts of central plaque lipid and necrotic inflammatory debris (15). Compared with asymptomatic carotid artery plaques, symptomatic carotid plaques are reported to have thinner, less fibrous caps, infiltrated with foamy macrophages and T cells that are more prone to rupture or ulceration (2,16), and to contain a central, lipid-rich necrotic core that may be closer to the cap (17). Several studies have suggested that the prevalence of intraplaque hemorrhage or thrombus adherent to the plaque surface is similar in symptomatic and asymptomatic carotid stenosis patients (2,18,–20), although one study found that fibrous cap thinning, plaque rupture, infiltration of the cap with foam cells, and intraplaque fibrin were more common in symptomatic than asymptomatic patients (20).
Inflammation and instability in recently symptomatic carotid plaques is strongly associated with macrophage infiltration, with plaque instability and rupture more frequently seen with a critical plaque cap thickness of <200 μm (16,21). Noninvasive carotid ultrasound studies have also suggested that symptomatic plaques tend to be more echolucent or hypoechoic, assumed to be secondary to a larger lipid-rich core, whereas asymptomatic plaques are more often echodense with smaller lipid cores on B-mode ultrasound (22,–24).
Although activation of the endothelium and coagulation system may contribute to pathogenesis of symptoms in patients with carotid stenosis (25,26), platelets also play a pivotal role in arterial thrombosis and thromboembolism. Exposure to high shear stress, as might occur in response to turbulent flow of blood through a stenosed artery, may activate platelets (27), promote platelet hyperreactivity, and potentially increase the risk of subsequent thrombosis and thromboembolism from a stenosing carotid plaque. Platelets may also play a role in the progression of atherosclerosis (28), and lipid-rich carotid atheromatous plaques may stimulate monocyte-platelet complex formation and platelet-dependent thrombus formation in vitro (29). Increased expression of the endothelial adhesion molecule, P-selectin (CD62P) (30), and platelet factor 4 (PF4) — a chemokine released by activated platelets (28) — in symptomatic vs. asymptomatic carotid plaques, suggests that persistent platelet activation may contribute to the evolution of atherosclerotic lesions.
To investigate the potential pathogenic role of platelet activation and function in patients with carotid stenosis and the potential for accessible platelet biomarkers to improve risk stratification of carotid stenosis patients, we performed a systematic review of the literature to critically assess available data on ex vivo platelet activation and platelet function/reactivity in blood in this patient population. The main aims of this review were to consolidate available data and to stimulate further research in this important area of translational platelet science, vascular neurology, and stroke medicine.
Methods
A systematic review of the literature was performed by searching PubMed, Medline, Ovid, Embase, and Web of Science/Web of Knowledge for human studies on platelet activation or platelet function in symptomatic or asymptomatic carotid stenosis published in the English language between 1975 and August 2011. The following search terms were used: ‘carotid stenosis’ and ‘platelet activation’, ‘platelet function’, ‘platelet reactivity’, ‘platelet aggregation’, and ‘flow cytometry’, We excluded case reports, case series, and review articles. One author (J. K.) personally read all abstracts and articles returned, hand searched the reference lists of published articles and relevant PhD theses in our department, and identified all papers suitable for inclusion in this review. After searching through all databases, 1467 articles and one relevant PhD thesis were retrieved. The articles were categorized a priori according to whether they compared data from patients with symptomatic or asymptomatic carotid stenosis vs. controls or other TIA or stroke sub-types, from patients with symptomatic vs. asymptomatic carotid stenosis, or from symptomatic or asymptomatic carotid stenosis patients undergoing intervention. After critical review of all manuscripts, 213 articles relating to assessment of platelet activation and/or function in patients with cerebrovascular disease were identified, and 26 were deemed suitable for inclusion in this systematic review. We subsequently categorized the evidence within these subject groups according to the method of assessment of platelet activation and function ex vivo.
Symptomatic carotid artery stenosis vs. controls
Platelet activation
Urinary (31) and soluble plasma markers of platelet activation (32,–38) have been studied in patients with symptomatic carotid artery disease. Another study assessed urinary markers of platelet activation in patients with carotid stenosis undergoing endovascular intervention, but symptomatic status or degree of stenosis was not mentioned (39).
Urinary markers
Urinary 11-dehydrothromboxane B2 (11-dehydro-TXB2) levels were increased in patients with chronic cerebral infarction with ‘distinct’ carotid atherosclerotic lesions (n = 6; degree of stenosis not reported) compared with controls (n = 44) or compared with patients with chronic cerebral infarction without distinct carotid atherosclerotic lesions (n=6; P≤0.01) (31). Another recent study found elevated creatinine-corrected urinary levels of 11-dehydro-TXB2 in subjects with carotid stenosis (n = 15) vs. healthy controls with no stenosis (n = 20; absolute values not given; P= 0.01) (39). These data do not inform us of the potential usefulness of urinary markers of platelet activation in patients with specific degrees of carotid stenosis.
Soluble markers (Table 1)
Symptomatic carotid stenosis vs. controls
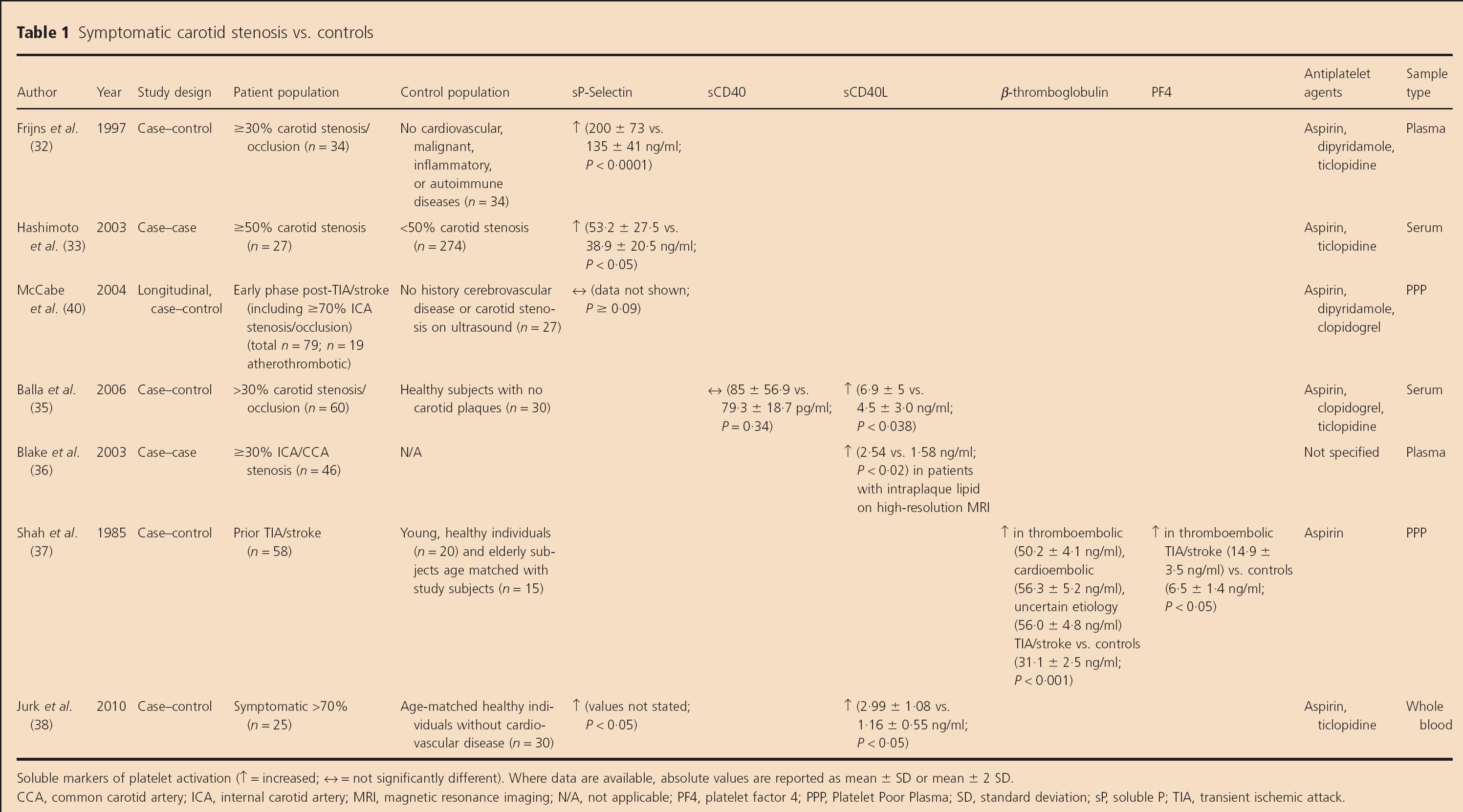
Soluble markers of platelet activation (↑ = increased; ↔ = not significantly different). Where data are available, absolute values are reported as mean ± SD or mean ± 2 SD.
CCA, common carotid artery; ICA, internal carotid artery; MRI, magnetic resonance imaging; N/A, not applicable; PF4, platelet factor 4; PPP, Platelet Poor Plasma; SD, standard deviation; sP, soluble P; TIA, transient ischemic attack.
Two studies found increased soluble P (sP)-selectin levels in patients with recently (greater than or equal to seven-days) symptomatic ≥30% carotid stenosis vs. controls without cardiovascular disease (n = 34 for each group, P> 0.0001) (32) and in symptomatic patients with ≥50% carotid stenosis compared with those with <50% carotid stenosis (n = 27 vs. 274; P < 0.05) (33). Although these data suggested that sP-selectin levels reflect enhanced platelet activation in recently symptomatic patients compared with controls, a further single center study did not reveal elevated sP-selectin levels in either the early or late phase after TIA or stroke onset associated with ≥70% carotid stenosis compared with controls (n = 19 early phase patients and 16 late phase patients vs. 27 controls, P ≥ 0.09) (40). It is important to note that sP-selectin is generated by platelets and endothelial cells, so it is not a specific marker of platelet activation (40).
Balla et al. assessed soluble serum CD40 and soluble CD40 ligand (CD40L) levels in 60 patients with carotid artery disease (16 ICA occlusions; 44 with >30% ICA stenosis) and 30 age- and gender-matched controls without carotid plaques (35). Soluble CD40 levels did not differ between patients and controls (P = 0.34), but CD40L levels were higher in patients than controls (6.9 vs. 4.5 ng/ml, P= 0.038).
Another imaging-based study demonstrated elevated soluble CD40L levels in 49 patients with ≥30% common carotid artery or ICA stenosis who had increased intraplaque lipid on high-resolution magnetic resonance imaging compared with those who did not have these plaque characteristics (P < 0.02) (36). Thirteen of these patients were ‘symptomatic’, but the proportion who were ‘recently symptomatic’ was unclear.
An earlier study demonstrated increased β-thromboglobulin (P < 0.001) and PF4 levels (P<0.05) in ‘thromboembolic TIA or stroke’ patients vs. controls; 10 patients (77%) had ‘ICA stenosis or occlusion’, but stenosis severity was not specified, and levels in the sub-group of patients with carotid stenosis were not specified (37).
A recent case-control study by Jurk et al. of 25 patients with acute (less than four-days) symptomatic >70% carotid stenosis, 48 patients with asymptomatic >50% carotid stenosis, and 30 age-matched healthy controls found that sP-selectin (P < 0.05) and CD40L levels were higher in symptomatic patients than controls (2.99 vs. 1.16 ng/ml; P < 0.05) (38).
Surface markers (Table 2)
Symptomatic carotid stenosis vs. controls
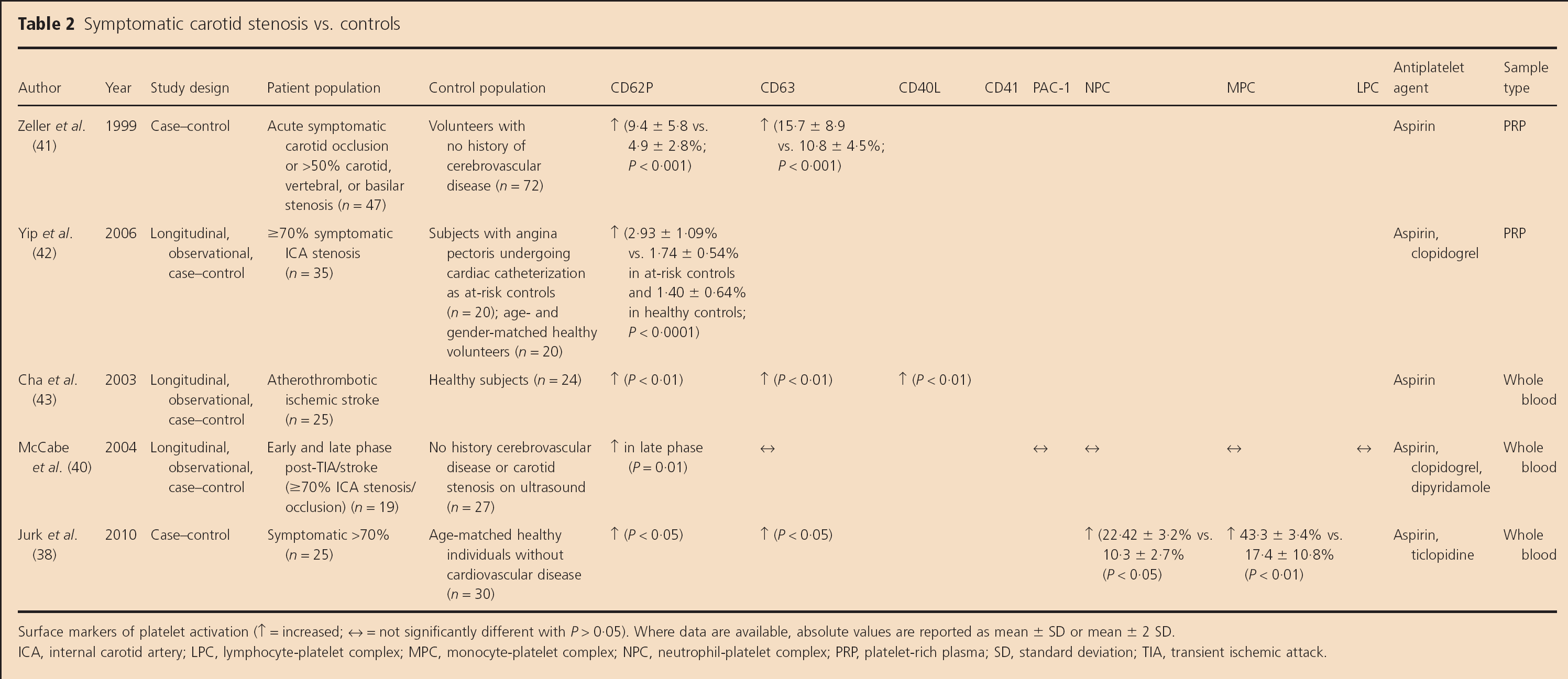
Surface markers of platelet activation (↑ = increased; ↔ = not significantly different with P > 0.05). Where data are available, absolute values are reported as mean ± SD or mean ± 2 SD.
ICA, internal carotid artery; LPC, lymphocyte-platelet complex; MPC, monocyte-plate let complex; NPC, neutrophil-platelet complex; PRP, platelet-rich plasma; SD, standard deviation; TIA, transient ischemic attack.
More recent studies have employed the sensitive and specific technique of flow cytometry to assess platelet activation [CD62P, CD63, and CD40L expression and leucocyte-platelet complexes (LPCs)] in platelet-rich plasma (PRP) (41,42) or whole blood (38,40,43,44) in symptomatic carotid stenosis patients vs. controls.
Zeller et al. found elevated platelet surface CD62P (P < 0.001) and CD63 (P = 0.003) in 47 patients within 36 h of symptom onset in association with symptomatic >50% carotid artery stenosis/carotid occlusion (n = 34; exact proportion with moderate or severe carotid stenosis alone not specified) and vertebral or basilar artery stenosis (n= 13) vs. controls (41). Yip et al. found elevated CD62P in patients at least two-months after TIA or stroke associated with ≥70% symptomatic ICA stenosis compared with ‘at-risk controls’ and ‘healthy volunteers’ (P< 0.0001) (42). Another whole blood flow cytometry study of 25 patients with acute (<24 h) ‘atherosclerotic ischemic stroke’ (proportion with >50% carotid stenosis not specified) found elevated platelet surface CD62P, CD63, and CD40L (P<0.01) levels compared with healthy controls (n = 24) (43). Sub-group data from a further longitudinal, case-control whole blood flow cytometry study revealed that CD62P levels were not increased within four-weeks but were significantly higher in patients with severe symptomatic carotid stenosis greater than or equal to three-months after TIA or stroke onset than in controls (P= 0.01) (40). However, the authors did not find higher levels of other platelet activation markers (CD63, PAC1, or LPCs) in patients than controls, although this study was not powered to look at individual TIA or stroke sub-types (40). Jurk et al. found that CD62P (P>0.05), CD63 (P>0.05), circulating monocyte-platelet (43.3% vs. 17.4%, P<0.01), and neutrophil-platelet complexes (22.4% vs. 10.3%, P<0.05) were elevated in recently symptomatic severe carotid stenosis patients vs. controls (38).
Platelet function (Table 3)
Symptomatic carotid stenosis vs. controls

Markers of platelet function and reactivity (↑ = increased; ↔ = not significantly different).
C-ADP, collagen-adenosine diphosphate; C-EPI, collagen-epinephrine; CT, closure time; CVD, cerebrovascular disease; PFA-100, Platelet Function Analyser-100; PRP, platelet-rich plasma; TIA, transient ischemic attack.
One pilot sub-study assessed ex vivo inhibition of platelet function with aspirin monotherapy on a platelet function analyzer, called the Platelet Function Analyser-100 (PFA-100), with both collagen adenosine diphosphate (ADP) and collagen-epinephrine (C-EPI) cartridges in 11 patients in the early phase (less than or equal to four-weeks) and nine in the late phase (greater than or equal to three-months) after TIA or stroke secondary to ≥70% carotid stenosis; data were compared with those from 23 healthy controls who were not on aspirin (45). Prior data from healthy controls have indicated that C-EPI closure times are increased in 83–100% of healthy controls on aspirin (46,–49). Median C-EPI closure times were not significantly prolonged in the early phase (P=0.3) but were prolonged in the late phase after symptom onset compared with controls (P< 0.001). Seven of 11 (64%) patients in the early phase and one of nine (11.1%) in the late phase had high ‘on-treatment platelet reactivity’/‘aspirin nonresponsiveness’ on the PFA-100 (45). However, six of these nine patients in the late phase had undergone carotid endarterectomy (CEA) or stenting before repeat testing, potentially explaining in part why the vast majority of these patients subsequently became more responsive to aspirin ex vivo.
Asymptomatic carotid artery stenosis vs. controls
Platelet activation
Three studies compared soluble markers (35,50,51), one compared surface markers (43), and two compared both soluble and surface markers of platelet activation (38,52) between asymptomatic carotid stenosis patients and healthy controls.
Soluble markers (Table 4)
Asymptomatic carotid stenosis vs. controls
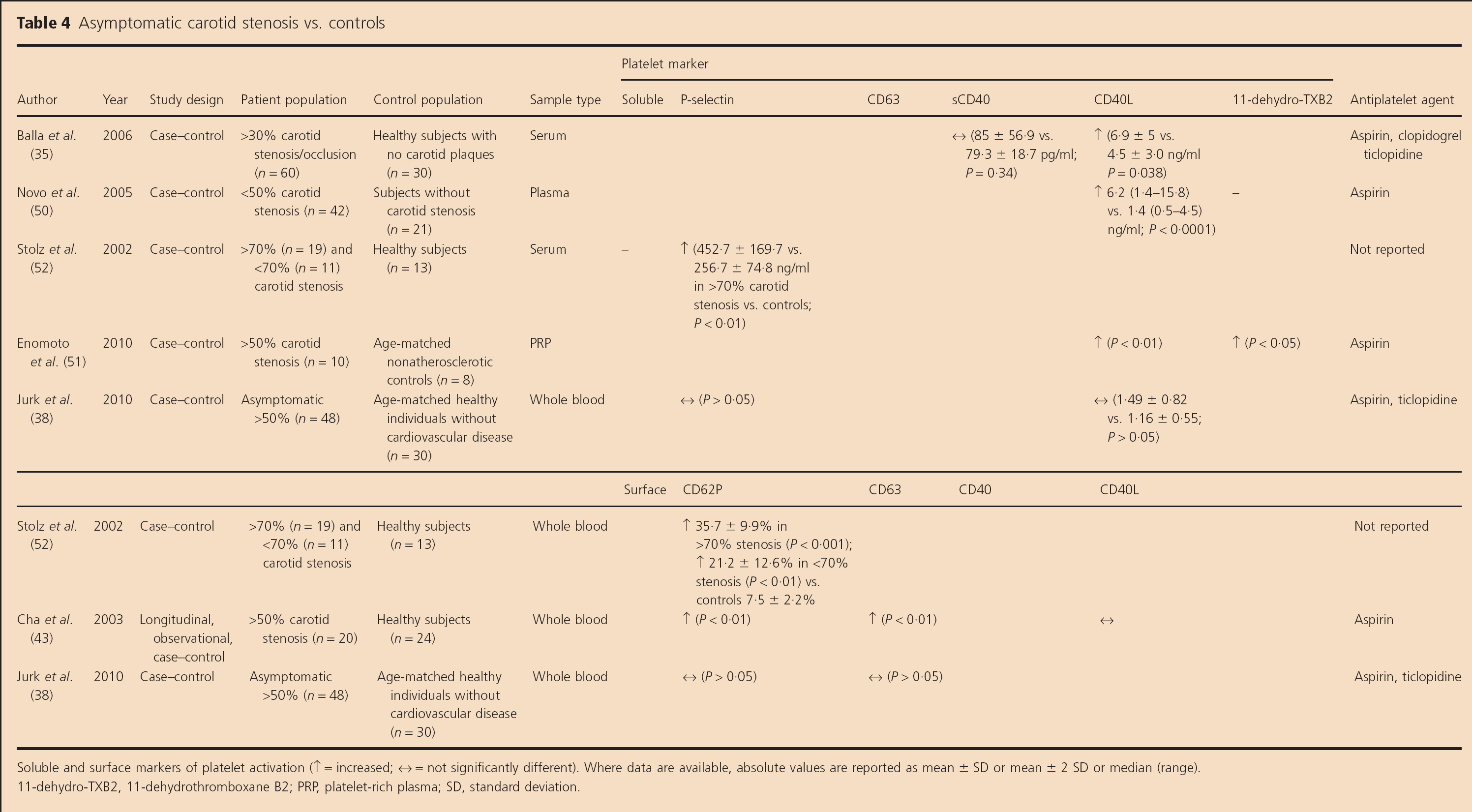
Soluble and surface markers of platelet activation (↑ = increased; ↔ = not significantly different). Where data are available, absolute values are reported as mean ± SD or mean ± 2 SD or median (range).
11-dehydro-TXB2, 11-dehydrothromboxane B2; PRP, platelet-rich plasma; SD, standard deviation.
Elevated soluble CD40L levels (P= 0.038) but not soluble CD40 levels (P=0.34) were identified in 60 patients with <30% carotid stenosis or occlusion who had not had a TIA or stroke within the preceding six-months compared with 30 healthy controls (35). Elevated soluble CD40L levels were reported in patients with <50% asymptomatic carotid stenosis compared with healthy controls (P < 0.0001) (50). Stolz et al. also found elevated sP-selectin levels in a pilot study of patients with asymptomatic >70% carotid stenosis compared with controls (P<0.01) (52). Another pilot study revealed elevated soluble CD40L (P < 0.01) and 11-dehydro-TXB2 (P<0.05) levels in PRP in 10 patients with >50% asymptomatic carotid stenosis vs. eight patients without carotid stenosis (51).
A further study did not reveal any difference in sP-selectin or soluble CD40L levels between asymptomatic carotid stenosis patients and controls (38).
Surface markers (Table 4)
In keeping with their pilot sP-selectin data outlined above, Stolz et at. found elevated platelet surface CD62P expression in 19 patients with asymptomatic >70% carotid stenosis (P < 0.001) and 11 patients with asymptomatic <70% carotid stenosis (mean stenosis severity 26 ± 20%; P<0.01) vs. healthy controls (n=13) (52). CD62P expression was also higher in patients with >70% than those with <70% asymptomatic carotid stenosis (P< 0.001). A whole blood flow cytometry study reported that patients with asymptomatic >50% carotid stenosis (n = 20) had higher CD62P and CD63 expression (P< 0.01), but not CD40L expression, than healthy controls (n = 24) (43). However, Jurk et al. did not find any differences in the expression of surface CD62P and CD63 between asymptomatic carotid stenosis patients and controls (38).
Symptomatic vs. asymptomatic carotid stenosis
One study compared soluble markers (38) and three studies directly compared platelet surface activation marker expression with whole blood flow cytometry in symptomatic vs. asymptomatic carotid stenosis patients (14,38,43).
Soluble markers (Table 5)
Jurk et al. found elevated sP-selectin levels (P < 0.05, absolute values not given) but not CD40L levels (2.99 vs. 1.49 ng/ml; P > 0.05) in symptomatic severe vs. asymptomatic >50% carotid stenosis patients (38).
Symptomatic vs. asymptomatic carotid stenosis
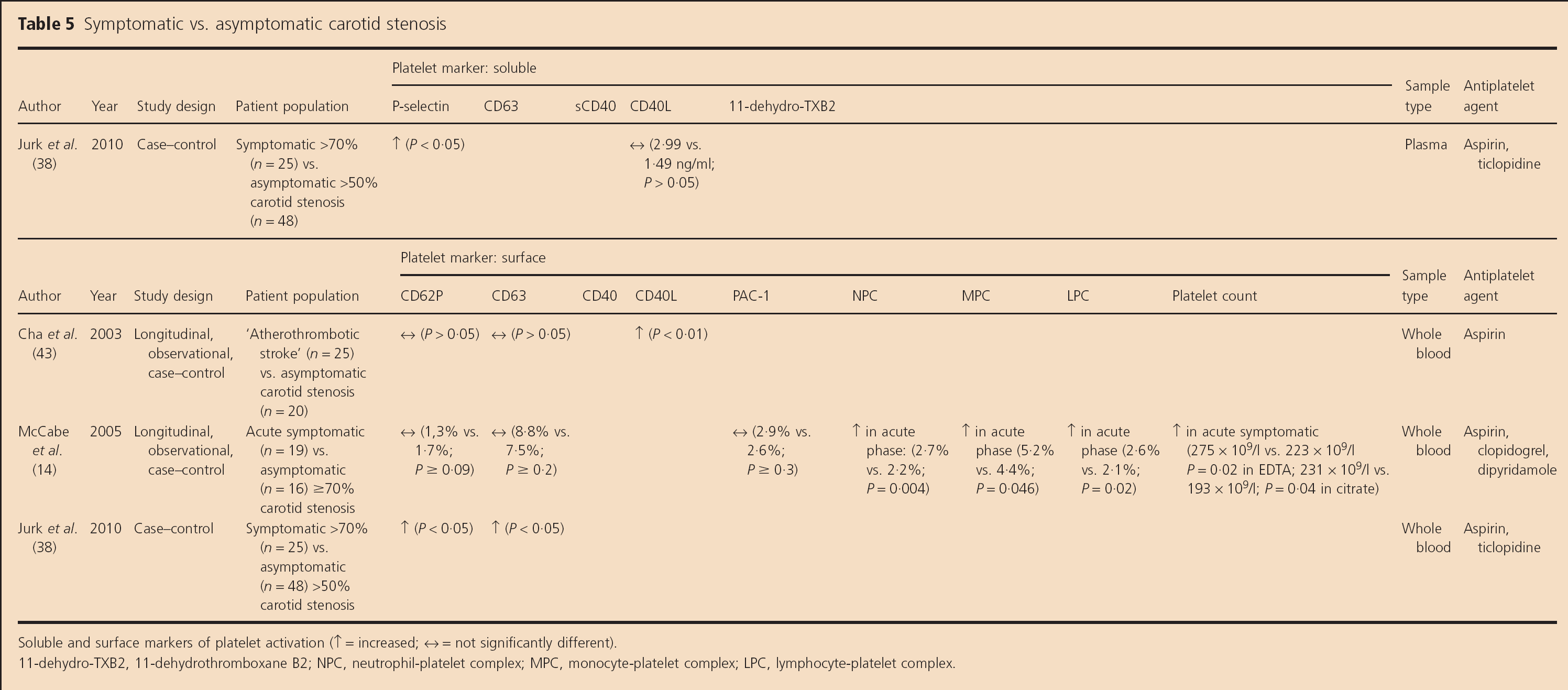
Soluble and surface markers of platelet activation (↑ = increased; ↔ = not significantly different).
11-dehydro-TXB2, 11-dehydrothrornboxane B2; NPC, neutrophil-platelet complex; MPC, monocyte-platelet complex; LPC, lymphocyte-platelet complex.
Surface markers (Table 5)
Platelet surface CD40L expression was found to be higher in patients with ‘acute large artery atherosclerotic ischemic stroke’ than in patients with asymptomatic carotid stenosis (P< 0.01), but there were no differences in CD62P or CD63 expression between the groups (43). However, the authors did not specify whether the symptomatic ischemic cerebrovascular event occurred in the territory of a carotid, vertebral, or intracranial stenosis.
Another pilot, longitudinal study identified an increased platelet count in both the early and late phases after TIA or stroke in patients with ≥70% symptomatic carotid stenosis compared with those with asymptomatic severe carotid stenosis (P ≤ 0.05); eight of 16 patients had undergone successful CEA or stenting by the late stage of follow-up (14). Circulating neutrophil-platelet, monocyte-platelet, and lymphocyte-platelet complexes were higher in the early (P ≤ 0.046) but not late phase after symptoms in symptomatic than asymptomatic patients (14). However, there were no differences in CD62P, CD63, or PAC-1 expression at any time-point between the groups.
Jurk et al. did find higher CD62P and CD63 expression in symptomatic severe compared with asymptomatic >50% carotid stenosis patients (P < 0.05; absolute values not given) (38).
Platelet function
There are no data directly comparing platelet function in whole blood in patients with symptomatic vs. asymptomatic carotid stenosis.
Symptomatic and asymptomatic carotid artery stenosis patients undergoing carotid or pharmacological intervention
Four studies assessed unstimulated platelet activation status (53,–56), and five assessed inducible platelet activation/reactivity with flow cytometry or platelet function testing in symptomatic carotid stenosis patients undergoing CEA (53,–55,57,58). Three studies assessed platelet aggregometry in carotid stenosis patients around the time of carotid intervention (55,56,59). Two recent studies assessed platelet aggregometry in symptomatic carotid stenosis patients after alteration of antiplatelet therapy (60,61).
Unstimulated platelet activation (Table S1)
Compared with levels at the time of initial skin incision and 1 and 24 h postoperatively, platelet surface CD62P expression in whole blood temporarily increased after carotid dissection (P<0.01) and immediately after cross-clamp removal (P < 0.05) in 40 carotid stenosis patients who underwent CEA on aspirin monotherapy; 90% were symptomatic, but the severity of stenosis was not specified (53). Platelet surface PAC-1 expression also temporarily increased after carotid dissection (P < 0.05) compared with levels following initial incision and 1 and 24 h postoperatively.
A further longitudinal study in patients with ≥70% symptomatic carotid stenosis found no differences in platelet surface PAC-1 binding or circulating monocyte-platelet complexes in the immediate precarotid stenting phase compared with three separate phases up to six-days after carotid stenting (44). A small perioperative study in symptomatic ‘high-grade’ carotid stenosis patients undergoing CEA found increased CD41 (P = 0.002) and CD62P expression (P < 0.001) during surgery, but no changes in LPC formation in the perioperative phase (54).
In a randomized controlled trial, Payne et al. assessed 100 carotid stenosis patients (mean degree of stenosis 80%; 84% symptomatic) immediately before CEA (55). All patients were stabilized on 150 mg of aspirin daily for four-weeks before intervention and were randomized to receive 75 mg of clopidogrel or placebo the day before surgery. Whole blood flow cytometry was performed on all patients prior to surgery, before and after addition of clopidogrel or placebo. There was no difference in platelet-fibrinogen binding between patients randomized to aspirin and clopidogrel vs. aspirin and placebo.
Vogten et al. assessed 27 patients who were scheduled to undergo CEA on either aspirin monotherapy (n=18) or aspirin and clopidogrel combination therapy (n = 9; stenosis severity and proportion of symptomatic patients not specified) (56). Platelet surface CD62P expression was assessed at seven time-points preoperatively, intraoperatively, and postoperatively; there were no differences in CD62P expression between patients on the two different antiplatelet regimens at any perioperative time-point.
Inducible and unstimulated platelet activation and platelet function (Table S2)
Assadian et al. assessed ‘inducible platelet activation’ (ADP- or thrombin receptor activating peptide-6 (TRAP-6)-induced platelet surface CD62P and CD41 expression), as well as unstimulated LPC formation and the numbers of reticulated platelets with flow cytometry, along with platelet reactivity on the PFA-100 in 20 ‘high-grade’ symptomatic ICA stenosis patients undergoing eversion CEA greater than or equal to three-months after symptom onset (54). Patients received either intraoperative unfractionated heparin (UFH) or enoxaparin. ADP-induced CD62P (P = 0.03) and CD41 expression (P < 0.001), and TRAP-6-induced CD41 expression (P = 0.002) increased during surgery (after administration of UFH or enoxaparin and after the anastomosis was sutured and declamped) compared with preoperatively and 30 min postoperatively. However, there were no differences between the sub-groups treated with UFH and enoxaparin. There were no differences in LPC formation between the immediate preop and postop phases in the enoxaparin and UFH groups combined. However, LPC formation was increased in the postop compared with the preop phase in the UFH sub-group, and this increase was higher in the UFH sub-group than in the enoxaparin sub-group (P = 0.034). There was a slight decrease in platelet count (P = 0.036) and an increase in reticulated platelets (P = 0.001) during CEA between cross clamping and re-establishment of blood flow through the artery. There were no differences in platelet reactivity on the PFA-100 between the immediate preoperative phase and up to 30 min after completion of the surgery (54).
Hayes et al. assessed ADP- and thrombin-induced platelet-fibrinogen binding with whole blood flow cytometry and ADP-induced platelet aggregometry in PRP in 120 patients with carotid stenosis (severity not specified) one-day prior to CEA; 90% of patients were symptomatic (57). Platelet fibrinogen binding was higher in patients with >25 vs. <25 emboli detected on three-hours of postop TCD ultrasound monitoring of the ipsilateral middle cerebral artery (P < 0.0001). The authors also found that ADP-induced platelet aggregation was greater in patients with >25 emboli than in those with <25 emboli detected on TCD following CEA (P = 0.0012) (57).
Robless et al. found elevated spontaneous (P < 0.001) and ADP-induced platelet aggregation (P < 0.01) in whole blood in patients at carotid clamping and after clamp release (P < 0.01) vs. initial incision; spontaneous platelet aggregation remained elevated up to one-hour after clamp release compared with levels at the time of initial incision (P < 0.05) (53).
A cross-sectional ex vivo platelet aggregometry study in 41 patients undergoing CEA for carotid stenosis (symptomatic status and degree of stenosis not clearly specified) found that administration of a heparin bolus immediately before CEA caused abrupt and significant elevations in the degree of arachidonic acid-induced platelet aggregation in PRP (P < 0.0001) vs. preoperative values before heparin administration. The authors postulated that this potentially explained why some patients are at increased risk of acute vascular events during CEA despite apparently appropriate preop aspirin therapy (58).
In the randomized trial alluded to above, the addition of clopidogrel to aspirin the day before CEA decreased ADP-induced platelet-fibrinogen binding on whole blood flow cytometry compared with treatment with aspirin alone (66.76% vs. 75.52%; P = 0.03) (55).
Szapary et al. assessed 18 patients with ≥70% carotid stenosis (10 with previous TIA or stroke and eight asymptomatic) before carotid endovascular treatment and found elevated ADP-induced platelet aggregation five-days following intervention compared with values measured immediately after the procedure (P < 0.0001) (59). However, no absolute values were provided by the authors, and the number of patients in this study was very small.
Platelet function after altering antithrombotic therapy in symptomatic and asymptomatic carotid stenosis patients
King et al. assessed platelet aggregation in PRP in patients with recently (less than one-month) symptomatic ≥50% carotid stenosis who were initially on treatment with aspirin monotherapy (61). Unmatched data from 30 patients subsequently randomized to receive clopidogrel (300 mg loading dose, followed by 75 mg daily) were compared with data from 30 patients randomized to receive 200 mg of dipyridamole modified release twice daily (MR bd) in addition to aspirin. ADP- and collagen-induced platelet aggregation were similar in both groups at baseline (P ≤ 0.83). ADP-induced aggregation was lower in the aspirin + clopidogrel than in the aspirin + dipyridamole group (86.5 vs. 101.7%; P < 0.001), but there was no difference in collagen-induced aggregation between the groups 48 h after commencing the relevant combination treatment (40.6 vs. 48.4%; P = 0.06). Longitudinal, matched data in each sub-group before and after changing treatment were not presented.
Payne et al. also performed platelet aggregometry in PRP in response to arachidonic acid in 100 carotid stenosis patients immediately before CEA, as outlined above (55). The addition of clopidogrel to aspirin the day before CEA did not significantly influence arachidonic acid-induced platelet reactivity compared with treatment with aspirin alone (3.8 vs. 4.3; P = 0.51).
Vogten et al. found that baseline arachidonic acid-induced platelet aggregation was higher in their carotid stenosis patients, described above, who were on aspirin monotherapy than in those on aspirin and clopidogrel combination therapy (14.5% vs. 10.3%; P < 0.05) (56). Five-minutes after administration of heparin and three-minutes before carotid artery clamping, arachidonic acid-induced platelet aggregation increased to 19.7% in the aspirin group and 22.5% in the aspirin and clopidogrel group vs. baseline preoperative arachidonic acid-induced platelet aggregation (10.7%; P < 0.01 and 7.5%; P < 0.05, respectively). In the 24-h period following surgery, platelet aggregation gradually returned to preoperative values in both groups. ADP-induced platelet aggregation did not change significantly in response to surgery or heparin administration.
The Clopidogrel and Aspirin for Reduction of Emboli in Symptomatic Carotid Stenosis Study was a randomized, double-blind study in patients with recently (less than three-months) symptomatic ≥50% carotid stenosis (61). All patients had TCD ultrasound monitoring, and if cerebral MESs were detected, they were randomized to receive either aspirin monotherapy or aspirin and clopidogrel combination therapy for seven-days. Collagen-induced platelet aggregation was performed in PRP at baseline and at day 7 following randomization. The ‘mean maximum intensity of platelet aggregation’ was 106.7% of baseline at day 7 in the aspirin monotherapy group (n = 40) and 70.9% of baseline in the aspirin and clopidogrel group (n = 31), implying that the addition of clopidogrel resulted in a significant reduction in collagen-induced platelet aggregation over aspirin alone.
Discussion
This review illustrates that relatively few studies have assessed platelet activation or platelet function/reactivity in patients with symptomatic or asymptomatic carotid artery stenosis. The existing data on indirect urinary markers or soluble plasma markers of platelet activation do not provide convincing, reproducible evidence of excessive platelet activation that is of potential clinical relevance to the modern-day management of patients with well-categorized asymptomatic and symptomatic carotid stenosis, in whom stenosis severity markedly influences treatment decisions.
Most, but not all, small-medium case-control studies included in this review have indicated that patients with mild, moderate, or severe symptomatic carotid stenosis exhibit increased platelet activation compared with healthy controls. However, only three studies (14,40,45) assessed the same symptomatic patients in both the early and late phases after TIA or stroke onset or carotid intervention, so it is unclear whether these findings are reflective of an acute phase response, whether increased platelet activation predisposes to TIA or stroke in patients with carotid stenosis, or whether there is a combination of both factors at play. Furthermore, many of these studies did not clearly define the exact proportion of symptomatic patients with ≥50% and ≥70% carotid stenosis in whom carotid intervention should be considered (31,32,34,36,37,41,43,54,57,58) or the interval between TIA or stroke onset and study inclusion (31,33,36,37,44,53,57,58,62). Allowing for these limitations, the majority of case-control studies included patients with symptomatic carotid stenosis already on antithrombotic therapy at the time of recruitment, thus indicating that platelets were excessively activated in these patients despite treatment with the secondary preventative regimen in question.
Within the symptomatic carotid stenosis sub-group, very preliminary data from one case-control study suggest that it is possible that surgical removal or endovascular treatment of a severely stenosing atherosclerotic plaque may reduce platelet activation and hyperreactivity ex vivo and decrease the likelihood of ex vivo aspirin nonresponsiveness/high on-treatment platelet reactivity in the laboratory at least three-months after intervention (45). However, these findings might also reflect resolution of the acute phase response following cerebral or ocular ischemia or infarction.
There is some evidence of enhanced spontaneous and inducible platelet activation and platelet hyperreactivity as measured by platelet aggregometry in PRP and whole blood in the perioperative phase around the time of CEA and also in ‘higher than lower risk’ carotid stenosis patients with more active embolization on TCD. These data provide some insight into the potential platelet-mediated mechanisms that may predispose to TIA or stroke around the time of CEA. However, these findings need to be assessed in larger patient cohorts, preferably in longitudinal studies, where patients in the acute phase following TIA or stroke are followed up to the late phase after intervention to assess whether changes in platelet reactivity persist over a more prolonged period.
Some case-control studies have revealed evidence of increased platelet activation in patients with asymptomatic carotid stenosis compared with healthy controls and that the degree of stenosis may influence the level of platelet activation. These data highlight the potential role played by ‘clinically silent’ carotid atherosclerotic plaques in contributing to platelet activation, although it is also possible that excessive platelet activation may contribute to plaque formation in this patient population in the first instance. Follow-up studies in asymptomatic carotid stenosis patients undergoing CEA are required to determine whether removal of the plaque may reduce platelet activation in these patients. However, due to the small sample size, heterogeneous severity of the degree of asymptomatic carotid stenosis between studies, and differences in vascular risks between patients and control subjects, additional work is required to reinvestigate platelet activation status in asymptomatic carotid stenosis patients on modern preventative regimens.
To our knowledge, there are only three studies comparing platelet activation in symptomatic vs. asymptomatic moderate or severe carotid stenosis (14,38,43). These pilot studies provide some evidence that platelet production is increased in the early and late phases after symptom onset (14) and that platelets are excessively activated in recently symptomatic compared with asymptomatic moderate or severe carotid stenosis and controls (14,38,43). Measurement of circulating LPCs was found to be a more sensitive method of detecting increased platelet activation than measurement of platelet surface marker expression in one study (14) and may partly explain the higher risk of recurrent vascular events in symptomatic than asymptomatic carotid stenosis patients over time. However, as per other sub-group data outlined above, adequately powered longitudinal studies simultaneously assessing platelet surface markers and LPCs are required to readdress this issue.
There are very limited longitudinal data on the impact of altering antiplatelet therapy on platelet activation and function in patients with asymptomatic or symptomatic moderate or severe carotid stenosis. The differences in methodology employed between studies do not allow one to make any definitive conclusions on this issue at present.
The data outlined in this systematic review highlight the fact that although this aspect of translational platelet science in cerebrovascular disease is relatively underinvestigated, excessive platelet activation and/or platelet hyperreactivity is likely to play an important role in the pathogenesis of first or subsequent TIA or stroke in carotid stenosis patients. However, larger studies in asymptomatic carotid stenosis and further studies comparing platelet activation and function in symptomatic vs. asymptomatic carotid stenosis are warranted to improve our understanding of the mechanisms responsible for TIA or stroke and to improve risk stratification in this patient population. If longitudinal studies in patients with asymptomatic carotid stenosis confirm that the sub-group of patients with the highest levels of platelet activation are at highest risk of subsequent cerebrovascular events, these patients could be targeted with more aggressive primary prevention regimens, with or without surgery. Furthermore, if one could identify symptomatic patients with excessive platelet activation or platelet hyperreactivity and prove that these patients are at highest risk of recurrent stroke or TIA than those with less marked activation or hyperreactivity, these patients could be treated with more aggressive secondary preventative medical treatment and undergo hyperacute surgical or endovascular intervention, as deemed appropriate. Longitudinal follow-up studies will also be needed to determine whether a sub-group of symptomatic carotid stenosis patients, who have less marked platelet activation, might be treated successfully with a comprehensive medical secondary prevention regimen and avoid undergoing surgical intervention altogether, with the attendant health and economic benefits.
Footnotes
Acknowledgements
Study design; collection, analysis, and interpretation of data; and manuscript writing: J. A. Kinsella and D. J. H. McCabe.
Data analysis and interpretation; critical revision of the manuscript for important intellectual content: W. O. Tobin and G. Hamilton.