
Abstract
The prevalence of randomized controlled trials (RCTs) performed without fully informed prospective consent from subjects is unknown. We performed this study to estimate the prevalence of high-impact RCTs performed without informed consent from all subjects and examine whether such trials are becoming more prevalent. We performed a systematic review of English-language RCTs published from 2014 through 2018 identified in Scopus and sorted to identify the top 100 most highly cited RCTs each year. Text search of title and abstract included terms randomized controlled or clinical trial and spelling variants thereof, and excluded metaanalyses and systematic reviews. We independently identified the most highly cited RCTs based on predefined criteria and negotiated to agreement, then independently performed keyword searches, read, abstracted and coded information regarding informed consent from each paper and again negotiated to agreement. No quality indicators were assessed. We planned descriptive qualitative analysis and appropriate quantitative analysis to examine the prevalence and characteristics of trials enrolling subjects with other than fully informed prospective consent. We find that 44 (8.8%, binomial exact 95% CI 6.5% to 11.6%) of 500 high-impact RCTs did not secure informed consent from at least some subjects. The prevalence of such trials did not change over the 5 years (OR=1.09, z=0.78, p=0.44). A majority (66%) of the trials involved emergency situations, and 40 of 44 (90.9%) of the trials involved emergency interventions, pragmatic designs, were cluster randomized, or a combination of these factors. A qualitative analysis explores the methods of and justifications for waiving informed consent in our sample of RCTs.
Introduction
Securing informed consent for clinical trials is an international ethical requirement. Nonetheless, ethical codes such as the Declaration of Helsinki1 and the Council for International Organizations of Medical Sciences Guidelines,2 as well as laws of many countries, recognize circumstances in which prospective informed consent may be unnecessary. The standards justifying the waiver of the general requirement for informed consent vary widely, as do the rules for carrying out waivers.3 Typically, exceptions have been recognized for research in emergency situations where patients are at risk of having diminished capacity and time may be of the essence. One systematic review examined trials in acute stroke, reporting waivers of written informed consent in 9 of 36 trials (25%).4
Other justifications for waiving consent typically focus on very low levels of risk and the impracticability of securing consent (or the impracticability of carrying out the research if consent is required).5 6 For example, it is often argued that research using cluster randomization makes consent difficult because, depending on the specific design, there may be little interaction between subjects and researchers, and subjects may have limited choice or ability to refuse because the intervention is determined for them by their inclusion in the cluster.7 A recent review of a random sample of 39 randomized controlled trials (RCTs) conducted across the world using cluster randomization testing individual-level healthcare interventions found that more than 20% of the trials waived individual informed consent.8
Most recently, numerous bioethicists and researchers have been advocating the concept of a learning healthcare system that would strive for continuous improvement in the delivery of care.9 10 This strategy relies on greater use of pragmatic and comparative effectiveness trials, which attempt to embed trial procedures as much as possible into routine clinical practice and often are focused on resolving latent uncertainties about optimal care. Many authors have proposed ways for streamlining or simplifying the processes of informed consent,11-17 and in the limit, propose waiving consent from subjects.18 A recent systematic review examined pragmatic and comparative effectiveness RCTs performed at least in part in the USA and first published in 2014 and 2017, finding that 23 (22%) of 103 trials were done with a waiver of individual informed consent (LY Lin, N Jochym and JF Merz, unpublished data, 2020).
While the rates of waiving consent in these reviews seem high, there are no data for comparison. Thus, we sought to identify a background rate of published, high-impact RCTs that do not secure prospective fully informed consent from patients or subjects (or their family members or legally authorized representatives (LAR)), to examine the prevalence of trials involving emergency research, pragmatic or comparative effectiveness designs, or cluster randomization, and to describe the range of methods used for enrolling subjects without consent and the justifications for not securing consent. We focused our inquiry on highly cited RCTs, conceiving that the prevalence of foregoing or modifying informed consent in important, well-designed, thoroughly performed, and influential research, if not representative of the greater universe of clinical trials, may reflect state-of-the-art methods and presage or foretell where trial methodology may be heading.
Materials and methods
We performed a systematic review of RCTs focusing on the top 100 most cited trials published each year from 2014 through 2018. We performed a search of the literature using Scopus (https://www.scopus.com/). Scopus was chosen because it enables sorting of publications by number of citations. We searched titles and abstracts for ‘randomized controlled trial’, ‘randomized clinical trial’, ‘randomised controlled trial’ and ‘randomised clinical trial’ and excluded ‘systematic review’, ‘metaanalysis’ and ‘meta-analysis’ to identify all RCTs published in English-language journals (see the online supplemental materials). We constrained results by publication year (2014, 2015, 2016, 2017, 2018) and then sorted annual results by number of citations received. We narrowly defined our search for RCTs to minimize the burden of manual selection, believing that papers not using the specific language would be less likely to be RCT reports. The abstracts of the top 500 trials from each year were exported into Excel.
Abstracts were reviewed independently by the 2 authors (RD, JFM). We included all RCTs in which people were subjected to randomly assigned interventions for evaluation of their effects on biomedical or behavioral outcomes. Trials were excluded from the sample if they were: (1) uncontrolled or non-randomized trials; (2) retrospective studies; (3) consensus/committee statements, guidelines, commentary, editorials, and letters; (4) secondary analyses or long-term follow-ups of RCT data, including embedded non-randomized studies of RCT subjects or subgroup analyses of RCTs; (5) reviews, including systematic reviews and meta-analyses; (6) conceptual or methods papers; (7) non-health papers, including those with no health outcomes, such as educational interventions carried out in schools; (8) interventions with healthcare providers that did not assess patient outcomes; (9) pilot, feasibility, explorative, and proof-of-concept trials, as self-characterized by authors; (10) unpublished reports and conference abstracts that were not later peer reviewed and published; or (11) protocol, rationale, and designs of trials. Each author identified between 110 and 120 RCTs, and the authors negotiated to agreement to identify the top 100 most cited RCTs from each year for full-text review.
Both authors independently performed keyword searches to locate relevant language (eg, consent, inform, written, waiver, ethic, brief, retrospective consent, delayed consent, deferred consent, Zelen,19 cluster randomized, exception from informed consent (EFIC), and mental health act), examined every paper, and summarized approaches to informed consent for the full set of 500 trials. Geographic location of trial sites was coded for all trials into the following categories: USA, North America (but not USA), Central and South America, Europe, Africa, Asia, and Australasia. Protocols and supplemental materials were examined as necessary to understand the role of consent in each of the trials. Differences were negotiated to agreement.
We identified trials for which fully informed, prospective consent was not secured from all patients or subjects. This includes trials in which prior consent is not secured, as well as trials in which prior consent is secured but any information that would normally be disclosed is modified or withheld, including studies involving deception or those using limited disclosures. We did not code oral consent processes as waivers unless the information disclosed was stated by authors to be abbreviated or brief. That is, we did not consider waivers of written documentation of informed consent to be waivers of informed consent. Further, we do not consider trials proceeding with prospective parental consent for children or surrogate consent from incapacitated patients’ LAR or family members to involve waivers.
Our intent is to describe the journal and year of publication, the locations of study sites, the populations studied, trial methods including pragmatic, comparative effectiveness, and cluster randomized, the disease or condition of interest, the intervention, a description of the approach to informed consent, and justifications for not securing prospective consent. Appropriate descriptive statistics were planned, including contingency tables and tests of proportions. All analyses were run in Stata V.12.1 (2012; StataCorp, College Station, Texas).
Authors were emailed to obtain clarifications or additional information for all trials in which consent processes were unclear. Querying authors for details about their published trials is considered non-human subjects research by the University of Pennsylvania Institutional Review Board. This systematic review was done without patient or public involvement.
Results
We performed the Scopus search on October 29, 2019. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses diagram is shown in figure 1.20 Our initial literature search identified a total of 70,458 papers. We sorted results in each year by the number of citations each paper had received, then reviewed 998 titles and abstracts to cull out at least 110 RCTs for each year, ensuring that we would agree on a set of 100 for each year. Overall, the authors were highly concordant in title and abstract review (94% agreement, kappa=0.90), with very high agreement identifying RCTs (97% agreement, kappa=0.95). We attempted to contact 34 authors for clarification and further details about their trials, of whom 26 (76%) replied.
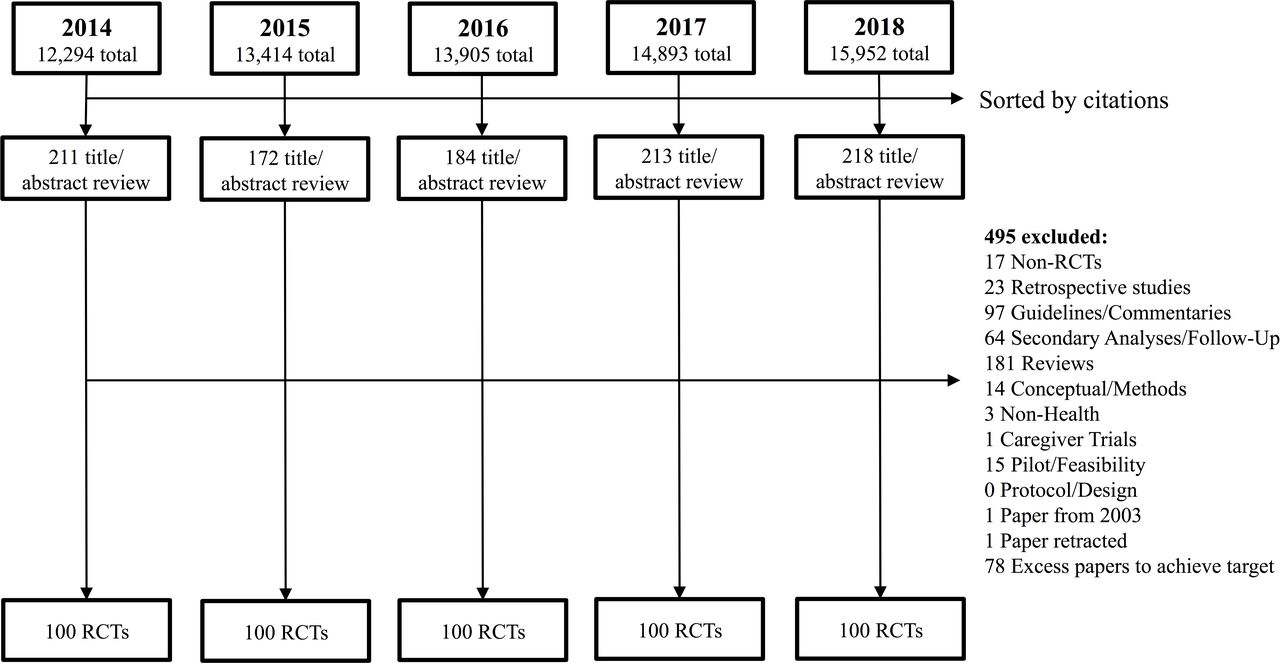
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) diagram. RCTs, randomized controlled trials.
Our review of all 500 papers yielded 44 trials (8.8%, binomial exact 95% CI 6.5% to 11.6%) in which prospective informed consent was not sought from at least some subjects. There were 8 such trials from 2014, 9 from 2015, 9 from 2016, 4 from 2017, and 14 from 2018. We ran a logistic regression to look for trend in likelihood of waiver by year, which shows no evidence of trend in our sample (OR=1.09, z=0.78, p=0.44). A complete list of all 500 RCTs is available from the authors.
We coded studies if the authors characterized the study as pragmatic trials (or involving pragmatic methods) or comparative effectiveness trials and if the trials used cluster randomization. For trials that used waivers, we coded whether authors noted emergent circumstances (eg, need for rapid treatment, emergency, and so on) as justifications for using waivers (see the online supplemental materials).
There were 52 author-described pragmatic trials or trials using pragmatic methods, 15 of which did not secure prospective consent from subjects (28.8%, binomial exact 95% CI 17.1% to 43.1%). There were 7 author-described comparative effectiveness trials, including 1 also described as pragmatic, and all secured prospective consent. Pragmatic trials in which fully informed consent was not secured are summarized in table 1.
Pragmatic trials that did not secure prospective informed consent from all subjects
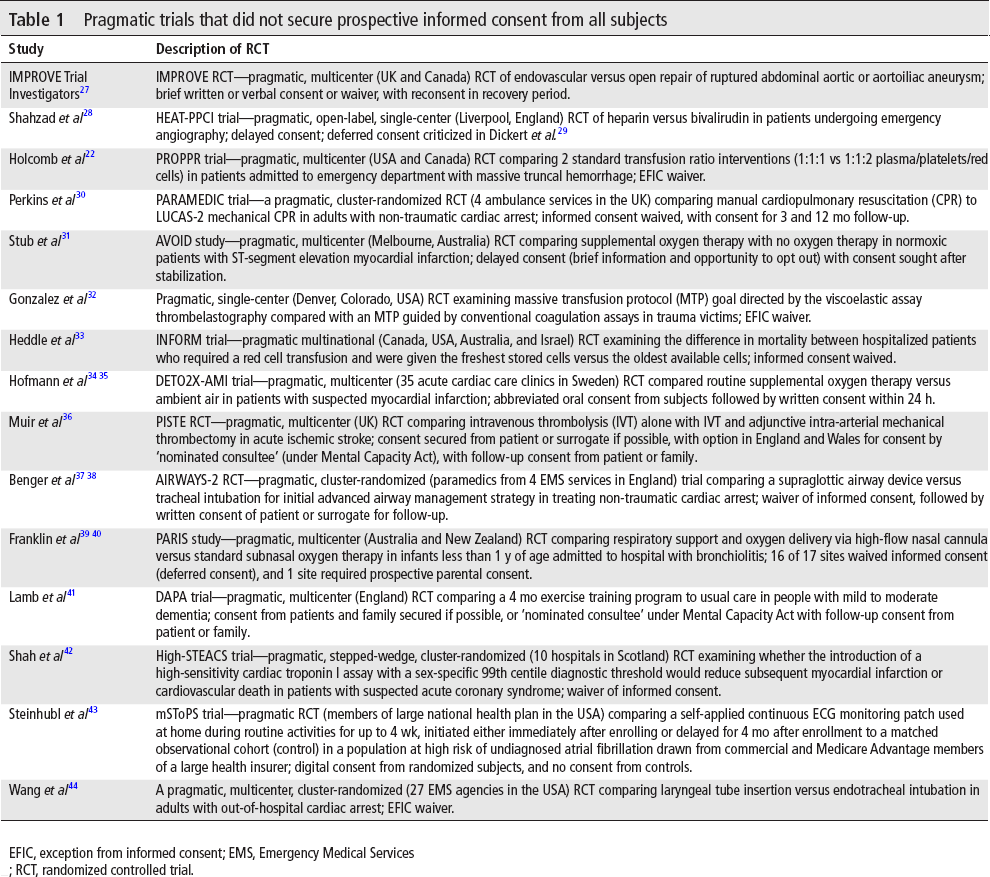
EFIC, exception from informed consent; EMS, Emergency Medical Services; RCT, randomized controlled trial.
Cluster randomization was used in 27 trials in the sample of 500, 11 of which did not secure consent from subjects (40.7%, binomial exact 95% CI 22.4% to 61.2%). Thirteen of the 27 cluster-randomized trials were published in 2018, suggesting that these methods might be becoming more popular. Notably, 6 of the trials completely waived consent, with no attempt to provide notice before, during, or after participation. Six self-described pragmatic trials used cluster randomization, 4 of which (66.7%, binomial exact 95% CI 22.3% to 95.7%) did not secure prospective consent from subjects. Non-pragmatic trials using cluster randomization are summarized in table 2.
Non-pragmatic trials that used cluster randomization
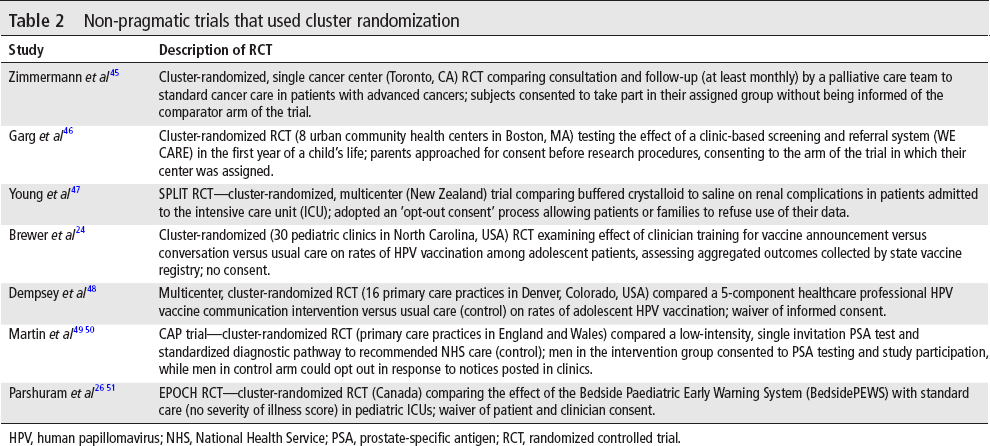
HPV, human papillomavirus; NHS, National Health Service; PSA, prostate-specific antigen; RCT, randomized controlled trial.
For the set of 427 trials that were neither pragmatic nor used cluster randomization, 22 (5.2%, binomial exact 95% CI 3.2% to 7.7%) did not secure fully informed consent from all subjects, and 18 of these involved emergency interventions. These trials are summarized in table 3. Of the 44 trials using waivers, 29 (66%) involved emergency circumstances, 11 of which were characterized as pragmatic trials, including 3 that were cluster randomized. Eighteen of the 29 were neither pragmatic nor cluster randomized. Overall, 40 of 44 (90.9%) RCTs in which fully informed prospective consent was not secured from all subjects were pragmatic, cluster-randomized, emergency research, or some combination of these.
Trials that were neither pragmatic nor used cluster randomization
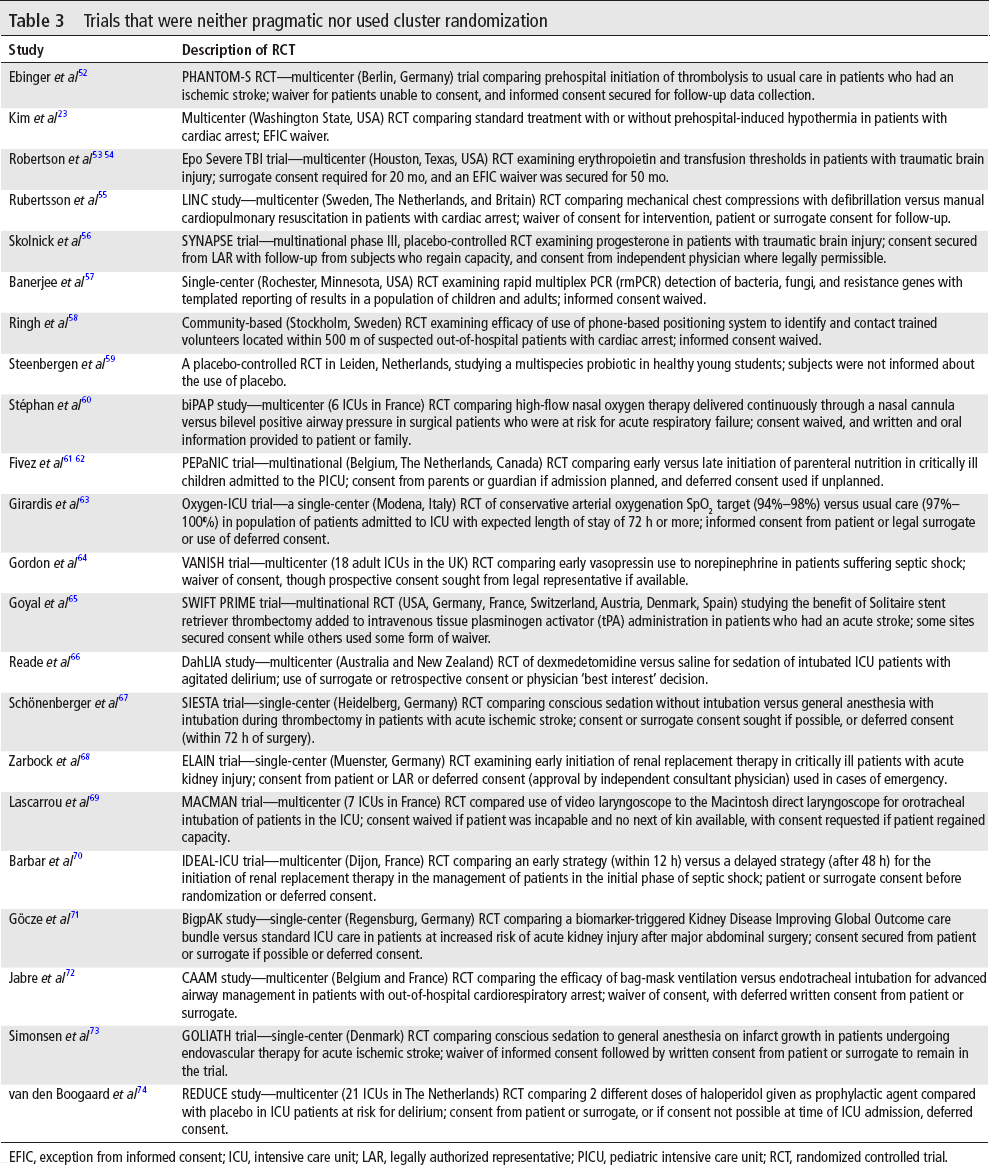
EFIC, exception from informed consent; ICU, intensive care unit; LAR, legally authorized representative; PICU, pediatric intensive care unit; RCT, randomized controlled trial.
Table 4 shows the journals in which the 500 RCTs were published, as well as the countries in which trials were performed. Not surprisingly, our sample drew heavily from journals having high impact factors. Overall, papers were published in 96 different journals, with 52 journals publishing only 1 of the trials and 18 journals publishing 5 or more of the trials. Table 4 also presents the number of trials published in those journals having more than 5% of the total sample (ie, 25 or more papers). The fraction of RCTs foregoing prospective consent to those securing consent published in the Journal of the American Medical Association (JAMA) and the New England Journal of Medicine (NEJM) is significantly higher than in the remainder of the sample by test of proportions (χ2=16.60 with 1 df, p<0.001).
Summary of number of trials and those that did not secure prospective fully informed consent
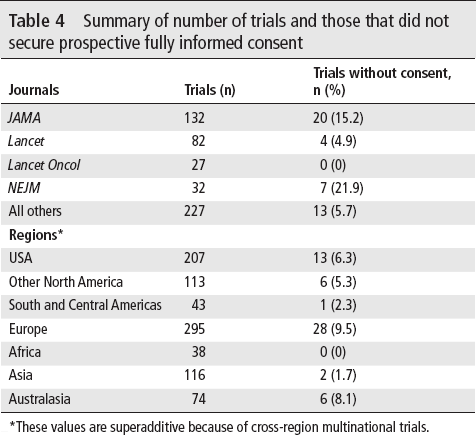
*These values are superadditive because of cross-region multinational trials.
A majority of RCTs in this review were performed with at least 1 site in Europe, followed by large pluralities in the USA, other North American countries, and Asia. Ninety-five trials were conducted solely in the USA.
Finally, we examined the explanations and justifications for not securing prospective consent either provided by authors or presumed based on trial details and regulatory requirements (details are provided in the online supplemental materials). Figure 2 maps these justifications against the methods of approaching consent, classifying the 44 trials. The figure shows that emergency research is the most common reason for foregoing consent (29 trials, 66%). The approaches ranged from completely waived consent with no notice or opportunity to opt out to, at the other end of the spectrum, attempts to secure consent from a patient if capable, or a family member or LAR if not, and otherwise deferring consent until some time after enrollment.
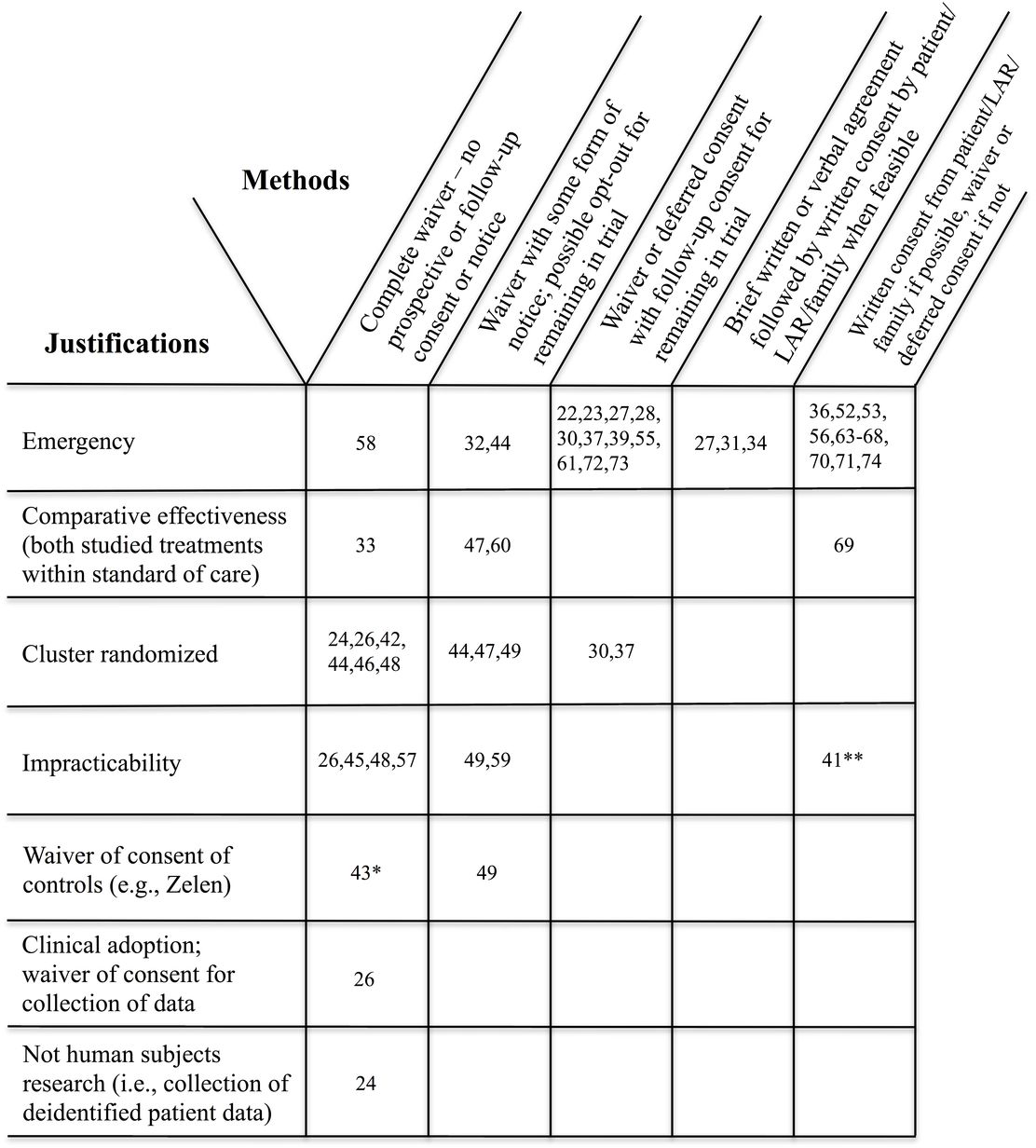
Reasons for not securing prospective consent from at least some subjects and methods of addressing consent in randomized controlled trials (RCTs). Numbers are reference numbers. When multiple references are provided for a trial, only the first reference appears in the figure. Note that trials may appear more than once.
Discussion
This is the first study to estimate the prevalence of published, high-impact RCTs that do not secure prospective informed consent from all subjects. Over the 5-year period, in a sample of the most highly cited RCTs published in the English language, 8.8% of 500 RCTs met our criteria for not securing prospective fully informed consent from all persons involved in the research. The majority of these trials (66%) involved emergent treatment, where patients often are incapacitated, time is of the essence, and family members who might consent may not be readily available. Nearly all of the trials (90.9%) in which prospective consent was not secured involved emergency situations, used pragmatic designs, or used cluster randomization, or a combination of these factors. This finding shows that the prior systematic reviews sensibly focused on emergency settings or trial design features that necessitated or were enhanced by provisions for waiving informed consent from at least some subjects. Nonetheless, a third of the trials were not emergency research, meaning that subjects from whom consent was waived had capacity and would be capable of providing consent, if informed and asked. The justifications for waiving or modifying consent vary widely, reflecting different national standards and accepted scientific practices.
The predominant approaches used in these trials involved either a waiver followed by securing patient or LAR consent after enrollment, or securing consent from a patient or LAR before enrollment, but if unable to do so, then waiving consent and securing consent when capable (or a LAR becomes available, where permitted). Some trials used a brief or abbreviated verbal or written consent prior to enrollment, followed by a fully informed consent after enrollment. The US Food and Drug Administration EFIC rule technically falls in the middle, requiring consent of a patient or LAR if capable and time permits or a waiver followed by notice and an opportunity to opt out.21 Several US trials using EFIC waivers in our sample appear to have gone further than required and secured follow-up consent when possible.22 23
Methods of approaching waivers of consent varied broadly in this sample of trials. Eleven trials involved a complete waiver of informed consent, with no notice provided to subjects that the study was being done and no provision of information afterwards about their participation. Most of these trials were cluster randomized, where institutional structures such as hospitals, intensive care units (ICU), clinical practices or healthcare providers, or time periods are the units of randomization, with all patients receiving the treatment assigned by the trial protocol to the cluster in which they appear for medical care. The justifications for using cluster randomization, the nature of the interventions, and the ability of individual subjects to opt out, vary widely.7 One trial, which did not mention consent at all, tested training of providers about human papillomavirus vaccination communication, and collected practice-level data on rates of vaccination of adolescent patients from a state registry.24 Thus, the intervention was directed to altering patient behaviors and outcomes, but did not fulfill the US definition of research involving human subjects.25 A different trial implemented a risk assessment tool in pediatric ICUs as part of clinical practice, with waivers of consent of patients’ parents as well as clinicians for collection of follow-up data.26
Practices vary around the need to follow-up with subjects to either give them an opportunity to opt out, or to consent either to continued participation or to collection of follow-up outcome information. Some studies sought consent, others provided information by posting notices or by verbal or written disclosure, and some did nothing. It seems there is no broad ethical consensus or standard for when it is appropriate to tell people they have been in research and how to do so.
Finally, many of the trials in our sample used multiple approaches to consent or waiver, driven either by national regulatory requirements or institutional ethics review requirements. Seventeen trials (39%) sought consent from patients when capable, or surrogates when not, at least at some sites or some of the time, while other trials secured consent from some subjects but not from others (eg, controls). To what extent enrolling vulnerable patients without prospective consent is justified when it is feasible to run trials with populations capable of consent (directly or by surrogates) is worthy of further research.
The main limitation on our review is that we focused on highly cited papers, which drew a large number of trials from high-impact journals (NEJM, Lancet, and JAMA). While our sample drew from 96 different journals, we find the odds of a highly cited paper published in JAMA or NEJM foregoing informed consent to be significantly higher than in other journals. We are unable to correct for the volume of RCTs published in these journals over the 5 years of this study, which might paint a different picture. Determining whether the rate of performance of RCTs without prospective consent is the same for a broader set of less influential trials requires further research.
In conclusion, we find that almost 9% of high-impact, international RCTs published from 2014 through 2018 did not secure informed consent from at least some subjects. Nearly all of the trials using waiver involved emergency interventions, pragmatic designs, were cluster randomized, or a combination of these factors.
Footnotes
Acknowledgements
The authors thank the study investigators who responded to our inquiries and provided detailed information about their trials, Maylene Qiu for assistance with the systematic search, and several anonymous reviewers for helpful comments.
Contributors
JFM conceived the idea for the study and performed the data analysis; the authors contributed equally to the design, search, reading, coding, checking, and interpretation of data, and the editing of the manuscript. RD wrote the first version of the paper, and subsequent edits were managed by JFM. Responsibility for the study is solely that of the authors.
Funding
The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests
JFM reports personal fees from Merck Sharp & Dohme and the American College of Radiology Imaging Network and a grant from Public Responsibility in Medicine and Research, all outside the submitted work.
Patient consent for publication
Not required.
Provenance and peer review
Not commissioned; externally peer reviewed.