
Abstract
Acromegaly is a rare disease associated with comorbidities that are common in the general population. Most patients undergo screening for classic phenotypical (CP) or mass effect manifestations. By retrospective review of pituitary tumor surgeries performed between 1994 and 2016 (1836), we identified patients with acromegaly (112). Main presentations were: CP (43%), mass effect (26%), incidentally detected (ID) tumors (17%), and other (14%). We compared the ID and CP groups regarding prevalence, clinical, biochemical, radiological and histopathological characteristics, and postoperative outcomes. The prevalence of ID among all surgeries increased after 2011 from 0.6% to 1.9% (p=0.01), while prevalence of CP remained stable (2.8% and 2.33%, p=0.65). Almost half of ID (47.4%) presented with otolaryngological manifestations. The ID and CP groups were similar regarding age, gender, comorbidities (hypertension, diabetes, hypopituitarism), tumor diameter and cavernous sinus invasion. Median insulin-like growth factor (IGF-1) and growth hormone (GH) levels were lower in the ID than CP (p<0.05 and p=0.07). Patients younger than 40 had smaller tumors in the ID than CP, while the opposite was true for older patients. The 3-month biochemical remission rates were similar (68% ID and 58% CP). A similar number of patients had normal IGF-1 at last follow-up (89.5% ID and 81.25% CP) after surgery alone and multimodality treatment. In conclusion, an increased number of patients with GH-secreting adenomas were ID in recent years. Education of physicians other than endocrinologists regarding presentation and comorbidity clustering may lead to an earlier diagnosis of acromegaly and improved outcomes.
Significance of this study
What is already known about this subject?
Acromegaly is a rare disease causing debilitating comorbidities and increased mortality, which correlate with the duration of uncontrolled growth hormone (GH) excess.
Acromegaly is suspected based on physical changes of acral enlargement and facial changes.
With the increased use of MRI, prevalence of incidentally detected pituitary tumors increased in recent years.
Published data on incidentally detected GH-secreting tumors are scarce and based on single case reports.
What are the new findings?
This is the first study examining the presentation and long-term outcomes of patients with incidentally detected GH-secreting tumors.
These patients represented 17% of those operated for acromegaly at a tertiary referral center.
Most patients presented in recent years and almost half underwent head imaging in context of otolaryngological manifestations.
While biochemical markers of disease were lower compared with patients with a classic phenotypical presentation, radiological tumor characteristics, comorbidities and postoperative course were similar.
Younger patients with GH-secreting incidentalomas had smaller tumors and higher remission rates.
Significance of this study
How might these results change the focus of research or clinical practice?
All patients with pituitary incidentalomas benefit from biochemical screening for acromegaly with an insulin-like growth factor 1 level, even if in absence of classic features of acromegaly.
Clinician education is necessary for earlier recognition of acromegaly, based on typical features and a cluster of clinical manifestations characteristic for this disease.
Our study opens new research avenues for delineation of clinical course and histopathological characteristics of incidentally detected GH-secreting adenomas in a multicentric fashion in a larger number of patients.
Introduction
Acromegaly is a rare disease with a prevalence of 30-100 per million,1 usually caused by a growth hormone (GH)-secreting pituitary adenoma. GH excess leads to the development of classic phenotypical (CP) features and manifestations: prominent brows, macroglossia, prognathism, enlargement of lips and nose, teeth shifting, acral enlargement and hyperhidrosis.2 Uncontrolled acromegaly is associated with metabolic, vascular, skeletal, cardiac and respiratory complications, which improve or resolve after biochemical control. Chronic GH excess leads to decreased life expectancy compared with general population.3 The diagnosis usually occurs 5-10 years after onset of manifestations, which causes long-term disability and decreased quality of life in a significant number of patients.4–6 Factors contributing to the delayed diagnosis are the insidious progression of physical features and the prevalence of ubiquitous comorbidities such as diabetes, obstructive sleep apnea, hypertension and osteoarthritis in the middle-aged general population.
With increased use of MRI for head imaging in the past 2 decades, more pituitary adenomas were incidentally detected (ID).7–9 A pituitary incidentaloma is defined by its serendipitous detection during imaging done for a reason unrelated to suspected pituitary pathology (14). There is a paucity of data regarding ID GH-secreting tumors. Few single case reports emphasized ID acromegaly in the setting of difficult endotracheal intubation or imaging for central nervous system conditions.10–12 In a study of 1692 patients operated for pituitary adenomas between 1999 and 2016, of the 52 incidentalomas only 1 had biochemical and immunohistochemical confirmation for acromegaly (prevalence 0.06%).13 Our group reported that changes in primary presentation of patients with acromegaly paralleled improved postoperative remission rates in recent years.14
Given its delayed clinical diagnosis and high morbidity and mortality, understanding the prevalence and characteristics of the ID GH-secreting tumors can lead to an earlier diagnosis of acromegaly. In the current study, we evaluate patients operated at our institution after ID presentation and compare their characteristics and postoperative outcomes with patients with CP presentation.
Materials and methods
Study design
We previously presented the methodology of data collection and case finding for this patient cohort.14 We extracted the cases of trans-sphenoidal surgery (TSS) by a single neurosurgeon (NMO) between 1994 and 2016 at Emory University Hospital in Atlanta, Georgia (GA), from the Research Electronic Data Capture database.
Clinical parameters
By retrospective chart review, we identified the primary presentation, that is, the reason for seeking medical care that resulted in the diagnosis of the pituitary tumor. We extracted information regarding specialty of physicians who started the pituitary work-up, comorbidities on presentation, physical features of acromegaly on physical examination at the initial evaluation at our center, as well as postoperative course and treatments.
Biochemical parameters
Diagnosis of acromegaly was based on elevated insulin-like growth factor 1 (IGF-1) level and abnormal GH suppression to oral glucose tolerance test. Biochemical remission was defined as normal IGF-1 and GH <1 ng/mL (random or after glucose challenge) at approximately 3 months postoperatively and during long-term follow-up.15 For patients in whom the GH data were not available and those treated with pegvisomant, IGF-1 alone was used to define biochemical control.
Imaging
MRI reports and neurosurgeon's notes were used to determine maximal tumor diameter and presence of cavernous sinus invasion (CSI). CSI was defined by tumor extension beyond the medial tangents of the 2 components of the intracavernous internal carotid artery.
Pathology
All tumors had confirmation of GH immunohistochemistry. Additional information regarding CAM5.2 immunohistochemistry and tumor proliferation markers was available since 2006 and 2011, respectively. Densely granular (DG) and sparsely granular (SG) patterns were determined by using mouse monoclonal antibody for CAM5.2.
Statistical analyses
We compared the ID and CP groups regarding preoperative characteristics and postoperative course. We ascertained differences between normally distributed continuous variables with t-test; for non-normally distributed variables, we used Kruskal-Wallis and Wilcoxon scores. For categorical variables, we used Fisher tests. We present the data as mean±SD for normally distributed variables and median (IQR) for non-normally distributed variables. Statistical significance was defined as p<0.05. Statistical analyses were performed with the SAS V.9.4 statistical software.
Results
Primary presentation for acromegaly
A total of 112 patients with acromegaly were operated between 1994 and 2016 by a single neurosurgeon (NMO) at Emory University Hospital in Atlanta, GA.
Patient groups were subdivided according to the reason for primary presentation that led to investigations that secured the diagnosis of acromegaly. Four groups were identified: CP changes, ID, mass effect group and ‘other’ (figure 1). The ID group consisted of patients who were diagnosed with acromegaly due to identification of a pituitary adenoma on imaging, which was done for reasons unrelated to a suspected pituitary lesion. The CP group consisted of patients who were suspected to have acromegaly in context of classic physical features (prominent brows, macroglossia, prognathism, enlargement of lips and nose, acral enlargement). The mass effect group comprised patients who had a work-up done for vision changes or headaches. The ‘other’ group included patients who had galactorrhea or hypogonadism and 1 patient operated in 1994 in whom information regarding presentation was not available.
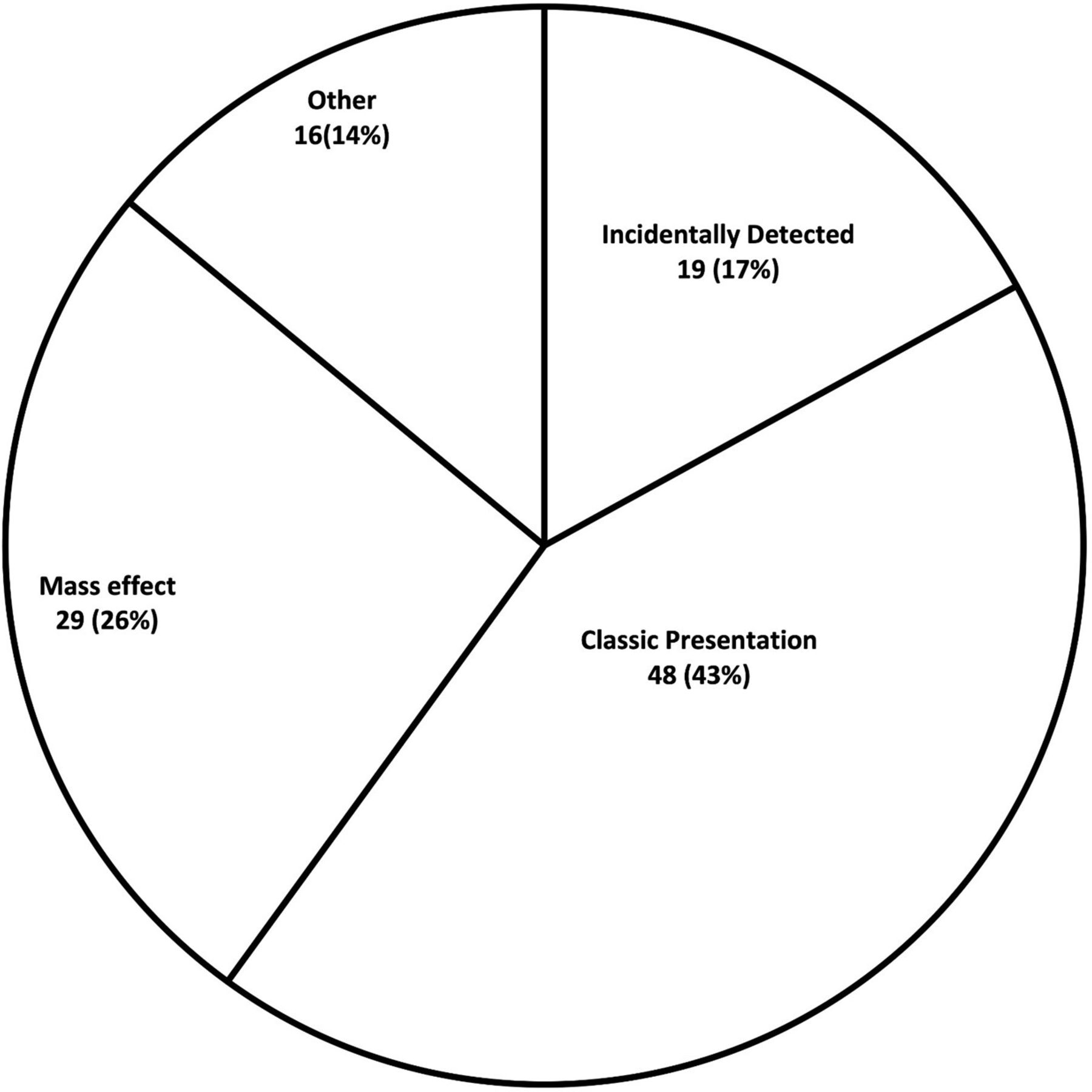
Primary presentation of 112 patients with acromegaly.
Prevalence of ID GH-secreting adenomas
Patients with ID GH-secreting adenomas (n=19) represented 1% of the 1836 patients operated by TSS for sellar and suprasellar masses during the study period (1994-2016). The first patient with GH-secreting incidentalomas was operated in 2007.
As previously reported, the 3-month postoperative remission rates in patients with acromegaly operated at our center increased after February 15, 2011 from 43.5% to 67.3% (p=0.01).14 After this date, only 30% of patients underwent screening for acromegaly for CP (compared with 54% previously). Instead, the proportion of patients screened for acromegaly in context of ID and ‘other’ (specifically hypogonadism) increased, and the proportion of patients presenting with neurological mass effect symptoms remained the same (figure 2).
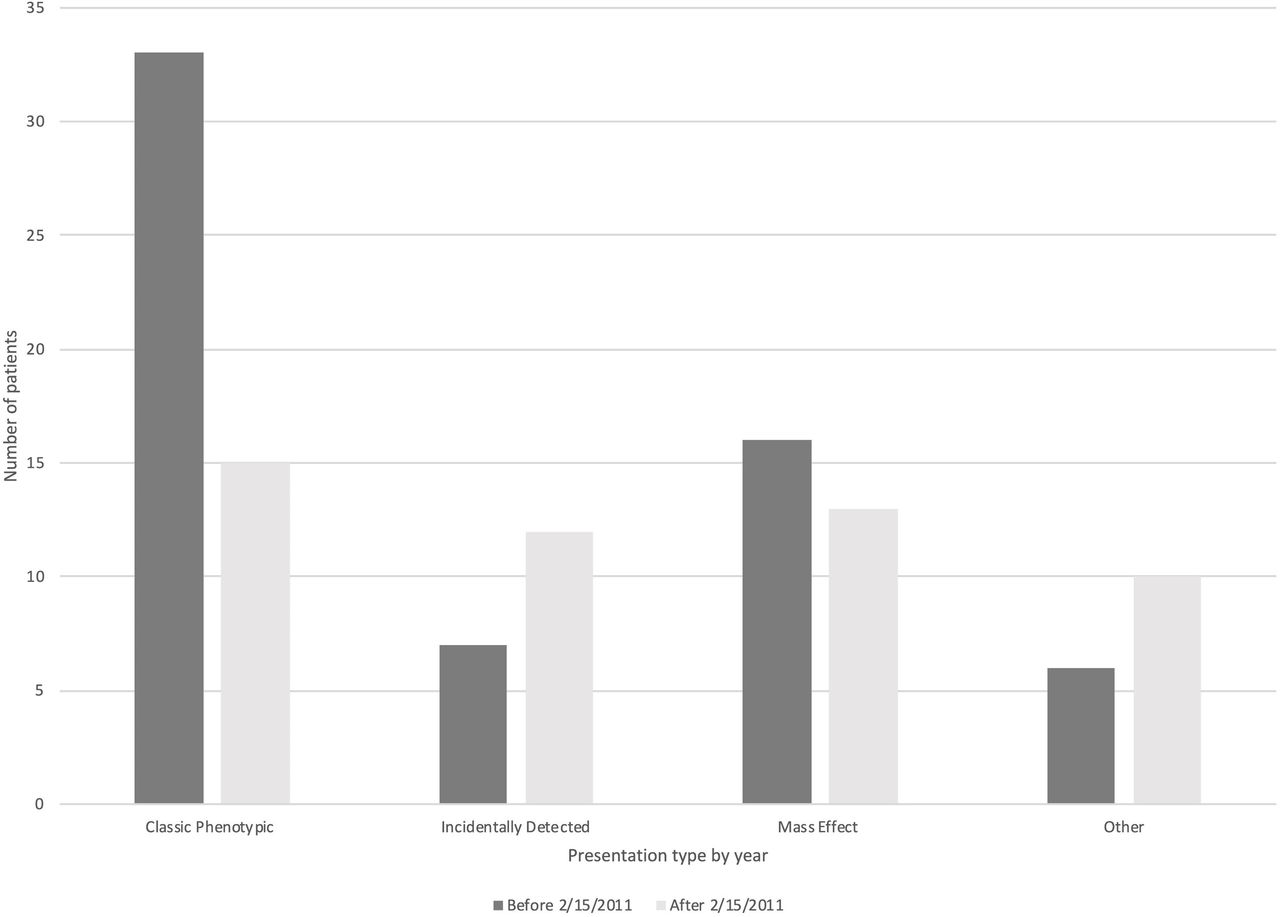
Changes in primary presentation in 112 patients with acromegaly operated over 22 y. The date February 15, 2020 was chosen because the 3 mo postoperative remission rates significantly improved after this date.14
The prevalence of ID GH-secreting adenomas increased from 0.6% before February 15, 2011 (1193 TSS) to 1.9% afterwards (643 TSS) (p=0.014).
Preoperative characteristics of patients with ID GH-secreting adenomas
In the ID group, almost half of the patients presented for ear, nose and throat (ENT) manifestations: hearing loss, sinusitis, voice changes, vertigo and tinnitus. The other reasons for head imaging are presented in table 1. The MRI that serendipitously discovered the pituitary adenoma was ordered by physicians with the following specialties: ENT (7), primary care (3), emergency room (3), neurology (2), oncology (2), urology (1), and endocrinology (1).
Patients’ characteristics and outcomes in the incidentally detected group
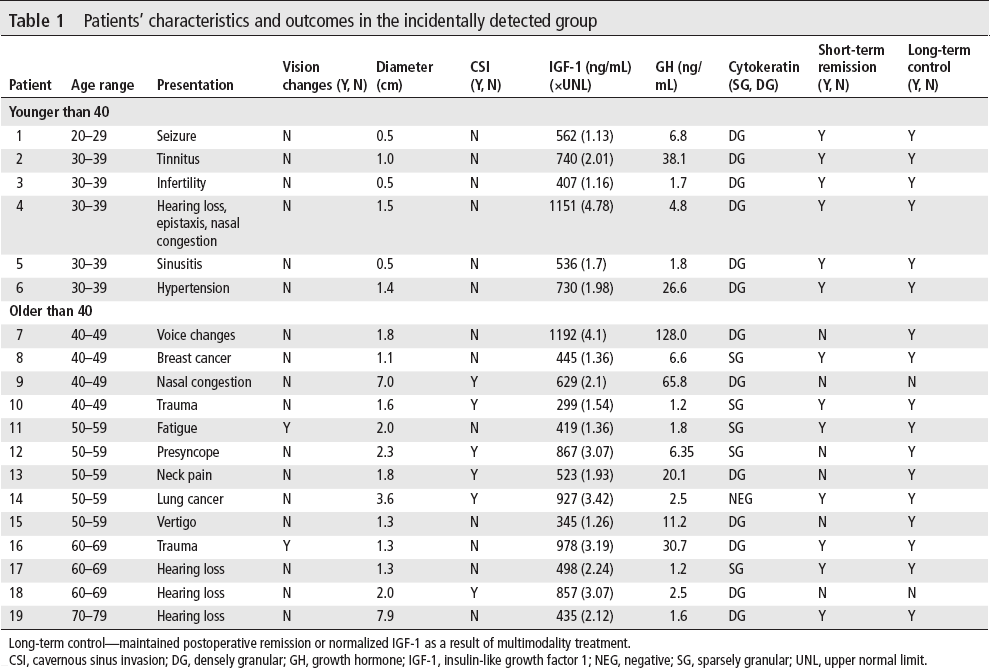
Long-term control—maintained postoperative remission or normalized IGF-1 as a result of multimodality treatment.
CSI, cavernous sinus invasion; DG, densely granular; GH, growth hormone; IGF-1, insulin-like growth factor 1; NEG, negative; SG, sparsely granular; UNL, upper normal limit.
Most ID patients had macroadenomas and almost a third had CSI on imaging. Despite incidental diagnosis, on evaluation at our center by pituitary specialists, the majority of patients had CP features of acromegaly. Preoperatively, comorbidities included diabetes mellitus in 5 patients (26.3%), hypertension in 11 patients (57.9%) and hypopituitarism in 5 patients (26.3%).
Postoperative outcomes of patients with ID GH-secreting adenomas
At 3 months postoperatively, biochemical remission occurred in 13 (68%) patients, and the tumor was no longer identified by imaging in 11 (58%) patients. Acromegaly-related comorbidities persisted in 4 (80%) of patients with preoperative diabetes mellitus, 10 (90.9%) of those with hypertension and 3 (60%) of those with hypopituitarism.
At end of follow-up of 2.92 years (IQR 2.9-4.0), 17 patients (89.5%) had normal IGF-1, which included patients who remained in remission postoperatively and those who achieved biochemical control with multimodality therapy. None of the ID patients in remission at 3 months postoperatively experienced biochemical recurrence. Of the 6 patients who did not achieve short-term remission, 3 had medical and radiation therapy, 2 medical therapy alone and 1 reoperation and medical therapy. Medical therapy consisted of somatostatin receptor ligands (SRL) in 4 patients, a combination of SRL and dopamine agonist (DA) therapy in 1 patient and a combination of SRL, DA, and pegvisomant in 1 patient.
Comparison between ID and CP presentation groups
Features of acromegaly on examination were present in all patients in the CP group as expected, but also in 58% of the ID group. Median IGF-1 and GH levels were lower in the ID than CP group (table 2). No differences regarding age, gender, comorbidities and imaging parameters were found between groups. However, there was a positive correlation between age and diameter in the ID which was not found in the CP group (figure 3).
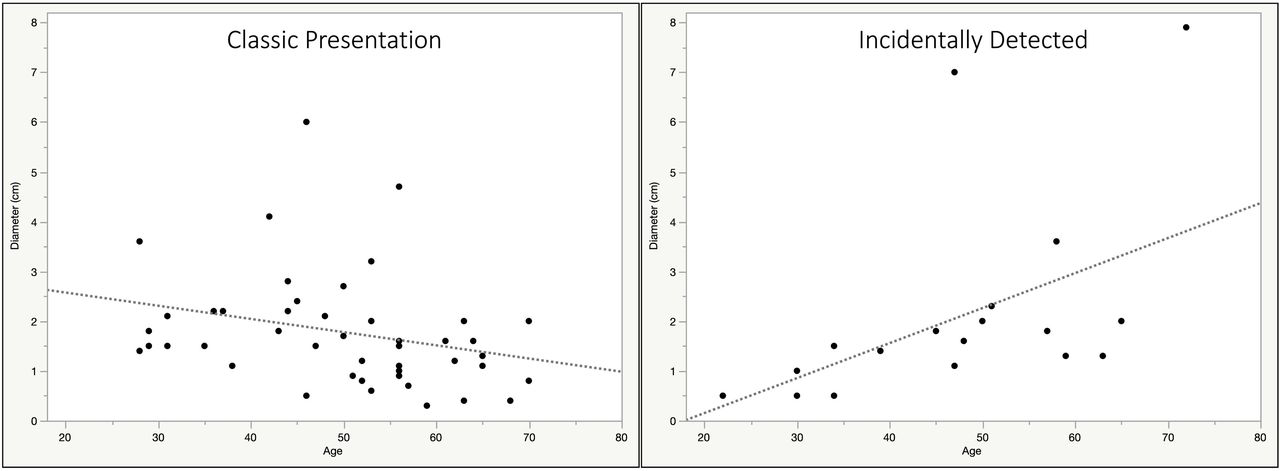
Relationship between age and tumor diameter in the incidentally detected (ID) and classic phenotypical (CP) groups. ID: R2=0.2, p=0.036; CP: R2=0.07, p=0.06.
Baseline characteristics of the 2 groups defined by incidental versus phenotypical changes presentation
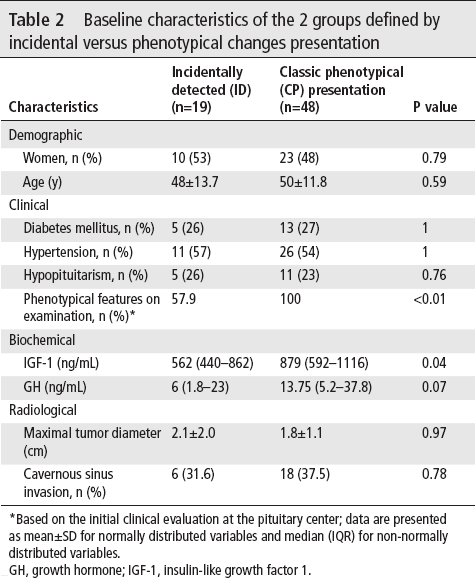
Based on the initial clinical evaluation at the pituitary center; data are presented as mean±SD for normally distributed variables and median (IQR) for non-normally distributed variables.
GH, growth hormone; IGF-1, insulin-like growth factor 1.
Prolactin immunohistochemistry (strong, moderate or weak) was found in 12/19 ID (63%) and 36/47 (76.6%) CP tumors (p=0.36). One tumor in the CP group did not have prolactin immunohistochemistry information available. Granulation status was available in 18/19 ID patients (13 DG tumors, 4 SG and 1 negative for CAM5.2) and 33/48 CP patients (20 DG and 13 SG). Although there were more DG tumors in the ID group (72.2%) than CP group (60.6%), this difference was not statistically significant (p=0.5). Histopathology evaluation of proliferation markers was available in 10/19 ID cases: 1 tumor had high Ki67 (7%-9%) and none had mitoses. In the CP group, 12/48 patients had proliferation markers available: 2 tumors had high Ki67 (3%-4%) and 1 tumor exhibited an increased number of mitoses without Ki67 elevation.
Short-term and long-term biochemical postoperative outcomes were not statistically different between the 2 groups. Remission at 3 months occurred in 13 (68%) patients in the ID group and 28 (58%) patients in the CP group. Biochemical recurrence was encountered in 0/19 ID patients and 3/48 CP patients. Approximately a third of patients required medical treatment in both groups, and a quarter required radiation therapy. At last follow-up, IGF-1 was normal in 80%-90% of patients in both groups as a result of surgery alone or multimodality therapy. All-cause mortality during follow-up was similar: 1/19 (5%) in the ID patients and 3/48 (6%) in the CP patients. Median follow-up in the CP group was 3.82 years (IQR 0.88-7.2).
Age subanalyses between ID and CP presentation groups
In the ID and CP groups, 6 (31.6%) and 10 (21%) patients respectively were younger than 40 (p=0.36).
In the ID group, the mean tumor diameter was smaller in younger versus older patients (0.9±0.5 cm vs 2.7±2.2 cm, p=0.01). Furthermore, none of the younger patients had CSI whereas 46% of the older ID patients did (figure 4). In the corresponding CP age subgroups, there was no difference regarding mean tumor diameter (1.9±0.7 vs 1.8±1.2, p=NS) or prevalence of the CSI (figure 4).
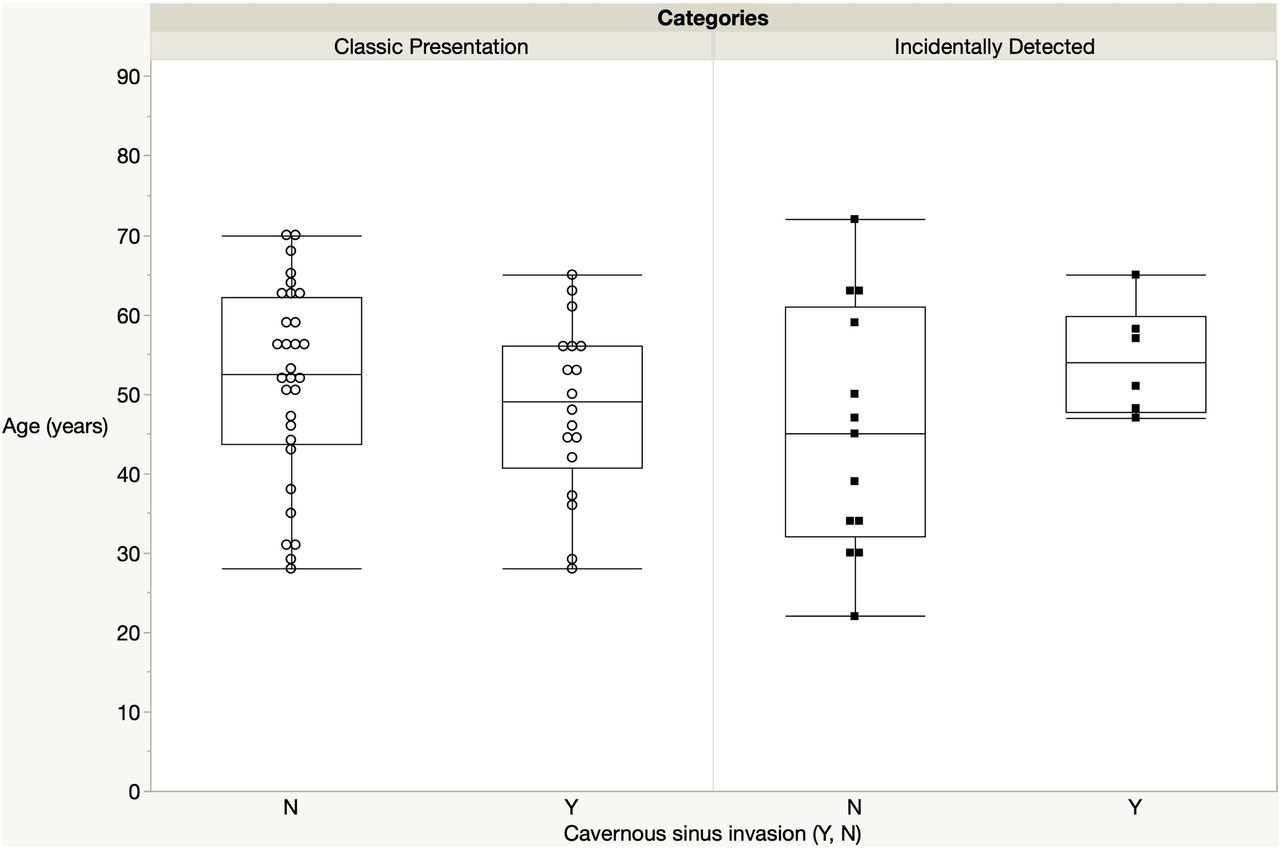
Relationship between age and cavernous sinus invasion in the incidentally detected (ID) and classic phenotypical (CP) groups. CP group: p=0.3; ID group: p=0.09.
When mean tumor diameter was compared between presentation subgroups by age, it was smaller in young ID than young CP (0.9±0.5 cm vs 1.9±0.7 cm, p<0.01). The opposite was true for patients older than 40, that is, larger mean diameter in the ID than CP (2.7±2.2 cm vs 1.7±1.2 cm, p=0.06).
On CAM5.2 immunohistochemistry, DG tumors were found in all adenomas from young ID patients and half of the older patients. Conversely, 4/7 young CP patients and 16/26 older CP patients with available information had DG adenomas. The small number of patients did not yield statistically significant subgroup differences.
All ID patients younger than 40 went into remission as a result of surgery alone and none experienced recurrence. In the CP group, 50% of young patients went into remission of whom 1 experienced recurrence. These differences were however not statistically significant.
Discussion
The study of patients with acromegaly serendipitously detected is relevant as it could reduce the time to diagnosis and the cumulative detrimental effects of high GH levels. To our knowledge, this is the first report on characteristics and outcomes of patients with GH-secreting incidentaloma from a surgical cohort. Almost 20% of biochemically and pathologically confirmed GH-secreting adenomas were ID between 1994 and 2016, with an increased prevalence of this presentation in the last 5 years of the study. ID patients had lower biochemical markers of acromegaly than those with CP presentation for acromegaly, but similar radiological tumor characteristics, comorbidities and postoperative course.
Epidemiological considerations
In our study of patients operated between 1994 and 2016, the first surgery for an incidental GH-secreting adenoma occurred in 2007, and the majority of ID patients were operated since 2011. The overall prevalence among all patients undergoing TSS during the study time frame was 1%, but increased from 0.6% before 2011 to 1.9% afterwards. An increase in the ID GH-secreting adenomas is in keeping with the demonstrated increase in pituitary incidentalomas in general in the last 10-15 years.7–9 While MRI was used since the early 1980s, 3T MRI has become available since the late 1990s,16 and has been incorporated into some practices since early 2000s.8 16 The increased use of head imaging has led to a higher incidence of pituitary incidentalomas. The majority of pituitary incidentalomas are small, clinically non-functioning and do not require surgery.15 However, the detection of GH-secreting incidentalomas has clinical implications, that is, treatment with surgery or medical treatment with improved long-term morbidity and mortality. In addition, ID GH-secreting tumors may affect the prevalence of acromegaly. Recent studies from Malta and Iceland have reported a higher prevalence of acromegaly of 125-137/million7 17 compared with 33-70/million in older studies from several European countries.18–20 The contribution of ID GH-secreting adenomas to a possible upward trend of acromegaly prevalence requires further study using national registries.
Diagnostic considerations
Information regarding how the patients with ID acromegaly come to medical attention is scarce and limited to case reports. Our study highlights that 47.4% of patients had imaging in context of ENT complaints. Indeed, acromegaly affects the pharyngeal morphology and causes uvula hypertrophy and macroglossia, in addition to nasal polyps and mucosal hypertrophy within the sinuses.21–24 A third of clinicians who ordered the initial head imaging study were ENT specialists, followed by primary care and emergency room physicians. Acromegaly is viewed as a rare disease characterized by classic physical changes in context of large invasive tumors which may not be amenable to complete resection. The authors emphasize that suspecting acromegaly should not be solely based on CP changes, but also when suggestive manifestations (excess sweating headaches, joint pain, sleep apnea, hypertension, hyperglycemia and hypogonadism) cluster together. Educational activities for medical students and residents should focus on its wide the spectrum at presentation and importance of early diagnosis. Among different specialties, our research indicates that patients sought advice from ENT and primary care physicians before endocrinologists.
When a pituitary tumor is detected serendipitously on head imaging, measurement of hormone levels is indicated even in absence of clinical features of hypersecretory syndromes. The Endocrine Society guidelines on pituitary incidentalomas in 2011 recommend screening for acromegaly with IGF-1 measurement.15
Clinical, biochemical and radiological considerations
Our study found that ID patients had lower median IGF-1 and GH levels but similar tumor size and prevalence of CSI as patients screened for acromegaly in context of CP. Lower IGF-1 levels in the ID versus CP group explain to some extent the incidental detection as clinical manifestations are linked to the degree of hormone excess. However, almost 60% of the ID patients had features of acromegaly when examined at our center. These patients could have potentially been diagnosed earlier. The authors underline the usefulness of old patient photographs during clinical evaluation, especially for patients with ENT complaints. Previous literature confirms that physical changes in acromegaly are often disregarded by both patients and physicians,6 although they are usually recognized by pituitary specialists.25
There were no differences between the CP or ID group regarding prevalence of diabetes mellitus, hypertension and hypopituitarism at presentation. While information regarding onset of comorbidities was not available, this suggests a similar temporal disease course in the ID and CP groups. This is further supported by a similar mean tumor diameter and prevalence of CSI between groups. Our group and others have shown a strong association of these radiological parameters with short-term and long-term postoperative biochemical outcomes.26–28 Our findings suggest patients with ID acromegaly have fewer physical changes in context of lower but sustained hormone excess.
Age considerations
In the ID group, we noted that younger patients had smaller tumors and there was a direct correlation between tumor diameter and age. This was not the case for the CP group. In addition, patients younger than 40 had smaller tumors in the ID than CP groups, while the opposite was true for older patients.
These findings suggest that patients incidentally diagnosed with acromegaly at a younger age have smaller tumors, corresponding to the natural course of the disease. Conversely, patients diagnosed based on classic features may present later in the disease course. While the number of patients in each age subgroup is small and our findings require further confirmation, our results have implications regarding postoperative outcomes, with young ID patients potentially amenable to complete resection and durable biochemical remission.
Limitations
The number of patients in the ID group is relatively small. Although this was sufficient for identification of statistical differences from the CP group, a larger number of patients are required to confirm our findings, especially for the age subanalyses. Another limitation is the retrospective nature of the study, with changing hormone assays and remission criteria over time and evolving WHO pathology recommendations. Fortunately, most patients had long-term follow-up which allowed for clarification of remission outcomes. The lack of updated pathology information regarding granulation status and proliferation markers in all patients did not allow for statistically meaningful comparisons between groups.
Conclusion
In summary, our study is the first report on presentation and outcomes of GH-secreting incidentalomas in a surgical cohort, which comprised almost 20% of operated patients with acromegaly. Our findings underline the importance of clinician and patient education to recognize acromegaly in early stages, thereby prevent the morbidity and mortality associated with advanced disease. The increasing incidence of ID tumors makes this relevant and timely, providing a window of opportunity for early intervention.