
Abstract

Plexiform angiomyxoid myofibroblastic tumor of the stomach, also termed plexiform fibromyxoma (PF), is a rare tumor of the stomach first reported in 2007 by Takahashi et al 1,2 Since that time, only 121 additional cases have been published in the literature. Due to the rarity, reporting the presence of this tumor allows for an increase in the knowledge of this disease process and a more robust literature review in the future.
The patient was a 39-year-old male who presented to his primary care physician for an unintentional 40+ pound weight loss over the last year. His physical exam was concerning for fullness in the right upper quadrant (RUQ). A computed tomography (CT) scan of the abdomen and pelvis was performed showing a hypoattenuated 4.8 × 5.9 × 5.6 cm solid mass in the RUQ that appeared to be arising from the gastric antrum (Figure 1). He was referred to surgery. A CT-guided needle biopsy was attempted but was undiagnostic. An esophagogastroduodenoscopy (EGD) with endoscopic ultrasound identified a mass in the antrum of the stomach. Under ultrasound guidance, biopsies were performed. The resulting pathology was suspicious for leiomyoma with immunostaining positive for Smooth Muscle Actin (SMA) and negative for CK117, DOG1, and CD45. A diagnostic laparoscopy was then completed to obtain a larger tissue sample using a Tru-Cut needle which confirmed gastric leiomyoma. Subsequently, a robotic-assisted laparoscopic gastric wedge resection of the antral mass with intraoperative EGD was performed. The patient’s postoperative course was essentially unremarkable, and he was discharged on postoperative day 1. The preliminary pathology identified a smooth muscle tumor of the wall of the stomach with myxoid changes. Immunostaining was positive for SMA and Desmin (DES) and negative for MUC4, C-Kit, DOG1, CD34, and S100. An outside tertiary institution’s second opinion was obtained. Final diagnosis was plexiform angiomyxoid myofibroblastic tumor of the stomach. At follow-up 1 month postoperatively, the patient’s symptoms have completely resolved with no evidence of recurrence. Computed tomography A/P showing a 4.8 × 5.9 × 5.6 cm mass in the right upper quadrant that appeared to be arising from the gastric antrum.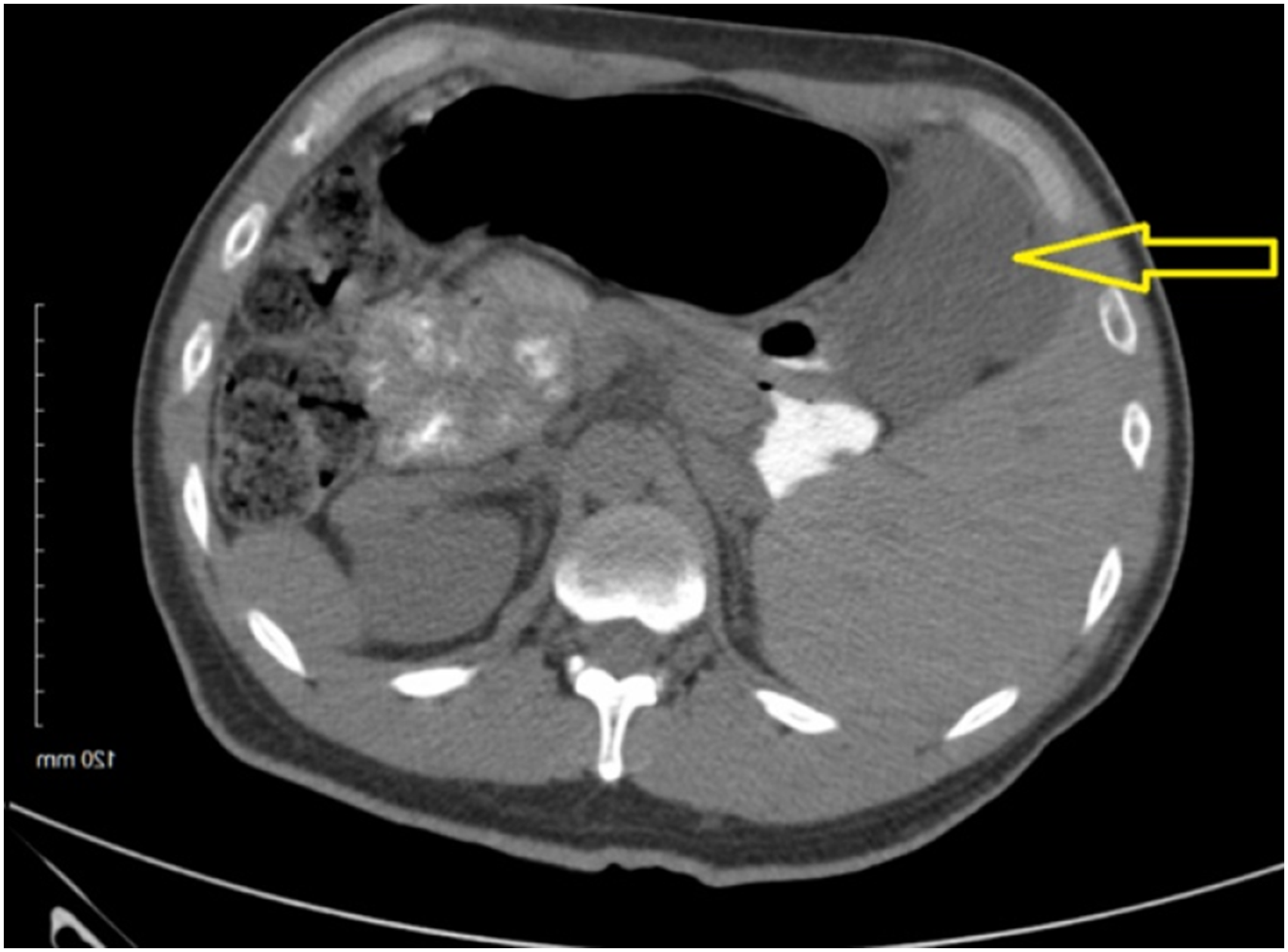
PF is a very rare benign mesenchymal tumor of the stomach first reported in 2007. 1 Obtaining an accurate diagnosis is imperative, as resection with clear margins appears to be curative. Imaging is the first step down the pathway of the correct diagnosis. PF occurs almost exclusively in the stomach in the antrum with sizes reported between 1.5 and 15 cm. PFs can have both solid and cystic components on CT but almost all are solid. A key distinguishing feature is mild enhancement of the solid portion in the arterial phase with strengthening enhancement throughout the venous phase. 3 There are many mesenchymal tumors of the stomach including gastrointestinal stromal tumors (GISTs), leiomyoma, schwannoma, and solitary fibrous tumor of the stomach. PFs may sometimes be confused with GISTs on imaging. GISTs commonly are a low-density solid tumor with hemorrhage and cystic change seen throughout the GI tract. Gastrointestinal schwannomas usually are solid and have homogenous attenuation. Once the mass is biopsied, histology with immunostaining confirms the diagnosis. Histologically, PF is described as having a delicate capillary network covering spindle cells growing in a plexiform pattern within myxoid or fibromyxoid stroma. This pattern helps distinguish PF from other mesenchymal tumors of the gastrointestinal tract such as myxoid GISTs, but further differentiation and confirmation of diagnosis come from immunostaining. 4 PF is positive for SMA, DES, and vimentin and negative for CD34, S-100, C-Kit, DOG1, and CD117. 4 This pattern of immunostaining was demonstrated in the patient reported here. Treatment is complete resection of the mass. An extensive literature review published in 2019 reporting on 121 patients demonstrated no local or metastatic reoccurrence of the 84 patients who followed up. 2 This demonstrates the curative nature with complete resection, but due to the lack of long-term follow-up of these patients’ jury is out on the metastatic potential of PF. The patient in this report at follow-up has yet to have any reoccurrence of disease. There are currently no follow-up recommendations in place for this tumor. Continued reporting of this as well as other rare tumors is imperative to allow for further understanding of the disease process leading to a more streamlined diagnosis and treatment plan.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.