
Abstract

Keywords
Introduction
Multiple endocrine neoplasia 2 (MEN2) is an inherited syndrome characterized by an autosomal-dominant transmission. It is further classified into 2 subtypes: MEN2A and MEN2B. Multiple endocrine neoplasia 2A is commonly associated with medullary thyroid carcinoma (MTC), pheochromocytoma, and primary hyperparathyroidism (PHPT), in order of frequency of clinical manifestation. This rare syndrome has an incidence of 2.8 per 100,000 live births and is caused by genetic mutations in the RET proto-oncogene, which is involved in cell growth and differentiation. 1
The diagnosis and treatment of MEN2A syndrome pose a significant challenge due to the wide range of organ systems affected as well as the severity and the age of onset of the different manifestations. Herein, we describe a case of sporadic MEN2A syndrome that required multiple surgical interventions and readmissions to a tertiary care center.
Case Report
This is a 26-year-old African American male with a significant past medical history of hypertension was referred after being diagnosed with primary hyperparathyroidism (PHPT) based on elevated calcium and parathyroid hormone (PTH) levels. The patient denied experiencing other classical symptoms of hypercalcemia or any familial history of endocrinopathies (thyroid, parathyroid, pancreas, or adrenal glands). As part of the investigation for hyperparathyroidism and the subsequent localization process, it’s part of our institution practice to obtain a four-dimensional computed tomography (4D CT) scan of the neck. This approach is preferred due to its higher accuracy compared to the sestamibi scan or ultrasound and is supported by our published experience.
2
(Figure 1) The scan revealed a 9 mm enhancing nodule located posterior to the left lobe of the thyroid, suggestive of an inferior parathyroid adenoma which correlated with the initial neck ultrasound obtained by the referring provider that showed a 9 × 5 × 3 mm solid hypoechoic nodule identified immediately inferior to the left thyroid lobe. There were no concerning thyroid nodules noted on this neck ultrasound. Consequently, this led to surgical exploration and parathyroidectomy. During the operation, a moderately enlarged left-sided parathyroid gland in the tracheoesophageal groove was identified and subsequently resected. However, PTH levels remained high, and further exploration revealed a metastatic papillary thyroid cancer to the central lymph nodes (based on initial examination by the pathologist), necessitating a total thyroidectomy and neck dissection. After extensive efforts to locate additional abnormal parathyroid tissue, none was discovered, resulting in termination of the procedure. Final pathology revealed multifocal medullary cancer in bilateral thyroid lobes; this measured 1 cm in its largest dimension with nodal metastasis to 2 out of the 6 lymph nodes that were obtained during surgery. Four-dimensional computed tomography of the head and neck demonstrating localization of parathyroid adenoma (red circles): (A) Axial view, (B) coronal view, and (C) three-dimensional reconstruction.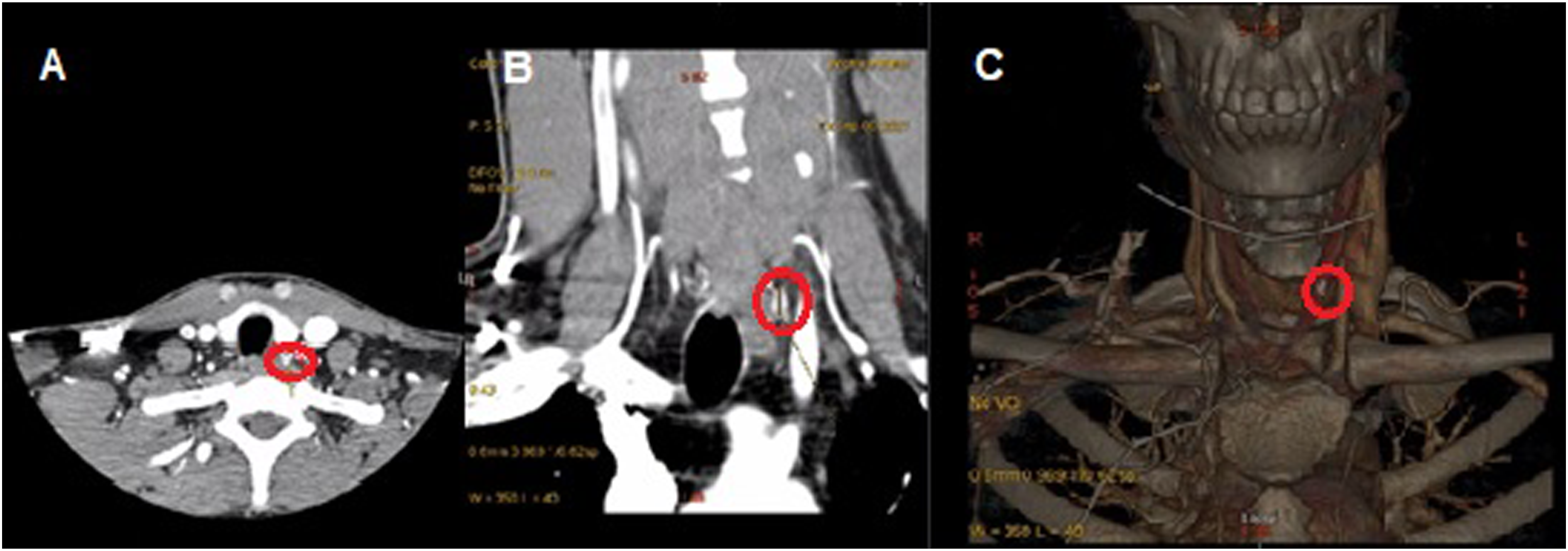
Postoperatively, a CT of chest and sestamibi scans were conducted to investigate the possibility of an ectopic parathyroid gland. The CT scan of the chest revealed an anterior mediastinal soft tissue mass, most likely residual thymus. Interestingly, bilateral adrenal masses were also observed in the upper abdomen, suggesting the presence of pheochromocytomas, and therefore multiple endocrine neoplasia (MEN2A) syndrome. Subsequent serum metanephrine levels of 409 pg/mL (normal range 0-88 pg/mL) and normetanephrines of 786.1 pg/mL (normal range 0-107.7 pg/mL) and a Ga-DOTATATE PET scan confirmed the presence of bilateral pheochromocytomas (Figure 2). The patient underwent bilateral adrenalectomy, followed by robotic video-assisted thoracoscopic parathyroidectomy with thymectomy to remove the ectopic parathyroid gland. Post-surgery, the patient was discharged with calcium carbonate (500 mg twice a day), calcitriol (1 mcg/daily), and magnesium oxide supplementation (400 mg twice a day) and hydrocortisone (20 mg in the morning and 10 mg in the evening) and fludrocortisone (.1 mg/daily). (A) Computed tomography of the abdomen showing bilateral adrenal masses (red arrows) and (B) Ga-DOTATATE scan showing high metabolic uptake of bilateral adrenal masses (red circles).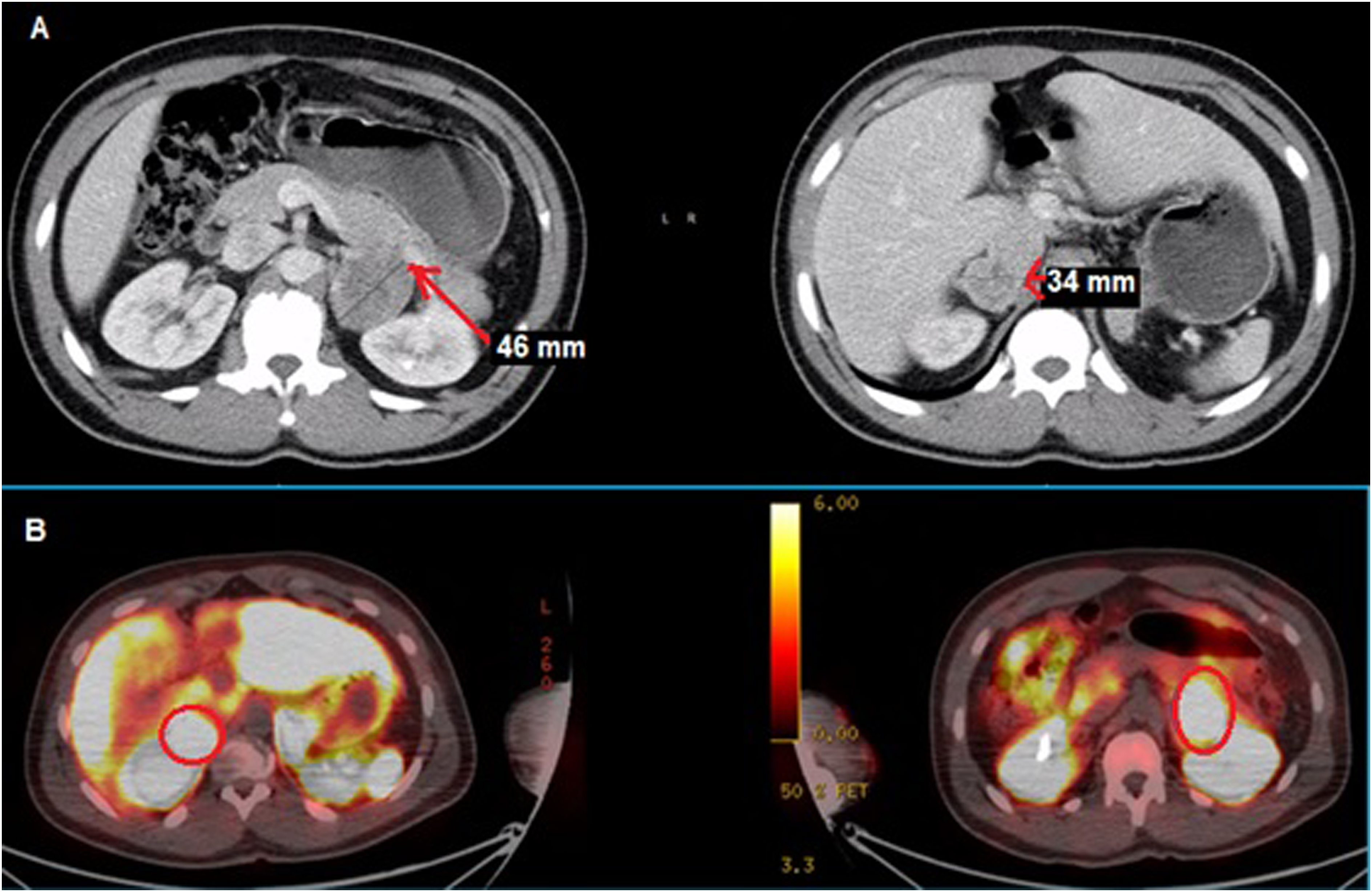
Despite initial stabilization of calcium levels, the patient faced challenges with critically low calcium levels, leading to multiple admissions to the hospital. After a period of stability, he experienced an adrenal crisis characterized by fever, low blood pressure, and increased white blood cell count. Empiric antibiotics and stress dose steroids were administered, leading to stabilization and subsequent discharge. He was discharged home with a higher dose of calcium carbonate (4 g/daily), and he was started on hydrochlorothiazide (12.5 mg/daily) per endocrinologist recommendations. Six months after surgery, the patient showed no signs of recurrent medullary thyroid cancer or hyperparathyroidism with calcitonin level of <2 pg/mL (normal range, <2 pg/mL), CEA of 2.2 ng/mL (normal range, 0-2.9 ng/mL), and calcium of 8.8 mg/dL (normal range, 8.2-10.4 mg/dL). His hypoadrenal state is well managed with supplemental adrenocortical hormones with the same dosing as we had previously prescribed. Subsequent genetic analysis revealed a mutation in the RET proto-oncogene.
Discussion
Most primary hyperparathyroidism (PHPT) patients exhibit a non-syndromic form, while over 10% of PHPT cases show an inherited syndrome. The occurrence of primary hyperparathyroidism (PHPT) as the initial manifestation of MEN2A cases is very rare, ranging from 0% to 11%. Most cases of index patients who present with PHPT as their first manifestation also have concurrent medullary thyroid carcinoma (MTC), often with lymph node involvement. 3 Even though the patient presented in this case did not have any history of a familial form of PHPT, the most recent consensus guidelines of the fifth international workshop published in November of 2022 state that genetic evaluation should be considered in patients <30 years old with PHPT, multigland disease, and those with family history of hypercalcemia or syndromic disease. 4
Conclusion
In summary, this sporadic MEN2A case underscores the complexities in diagnosing and treating patients with initial non-syndromic PHPT. Post-treatment, managing various neoplasms, alongside inpatient care for acute adrenal insufficiency and calcium imbalances, suggests the need for stricter follow-up and monitoring protocols to prevent such complications.
Footnotes
Author Contributions
VRS and JJ wrote and edited the manuscript. GM and AC edited the manuscript and approved the final version.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.