
Abstract
Primary hepatic neuroendocrine tumors (PHNETs) are exceptionally rare, posing significant challenges in diagnosis and management. To address these challenges, this study establishes an actionable, surgeon-facing decision framework that transforms clinical evidence into a stepwise management algorithm. By synthesizing evidence from the Surveillance, Epidemiology, and End Results (SEER) program (n = 446) and clinical series, we propose a structured pathway that explicitly links diagnostic confirmation, biological characteristics, and therapeutic strategy. The framework first necessitates the definitive exclusion of extrahepatic primary lesions through functional imaging (eg, PET-CT) and origin-specific markers to confirm the diagnosis. Subsequent operative decision-making is guided by biological assessment; higher tumor grades (G3: HR 2.94; G4: HR 3.04) are strong independent predictors of poorer survival, warranting a shift toward systemic control for high-grade disease. For localized disease, surgical resection remains the cornerstone of management, offering 5-year survival rates of 60-80%, whereas liver transplantation is prioritized as a viable curative strategy for unresectable, liver-confined G1/G2 tumors (5-year survival 70-80%). In advanced or multifocal stages, locoregional approaches provide effective palliation. This structured decision framework enables practicing surgeons to determine the optimal intervention—whether resection, transplantation, or palliation—based on individualized tumor grade, burden, and distribution, thereby optimizing long-term outcomes for this rare malignancy.
Keywords
Introduction
Neuroendocrine tumors (NETs), including carcinoid tumors, are heterogeneous groups of neoplasms that originate from the neuroendocrine system. Over the past few decades, the incidence of NETs has increased, and these tumors now account for approximately 1-2% of all gastrointestinal malignancies.1,2 The gastrointestinal tract remains the most common site for NETs, with 54.5 to 75% of cases occurring in the digestive system. Specifically, the small intestine (30.8%), rectum (26.3%), colon (17.6%), pancreas (12.1%), and appendix (5.7%) represent the most frequent sites of involvement.3,4 The liver is the most common site of metastases.3,4
In contrast, primary hepatic neuroendocrine tumors (PHNETs), however, are rare, comprising approximately 0.3% of NETs. 5 Due to the rarity of PHNETs and the challenge in distinguishing them from other liver malignancies, there is limited understanding of their origin, occurrence, development, clinical manifestations, diagnosis, and treatment. Early-stage PHNETs typically lack specific clinical symptoms. A small number of individuals may experience carcinoid syndrome, including symptoms such as skin flushing and diarrhea, whereas others may be identified incidentally during routine physical examination. 6 Advanced PHNETs more commonly cause symptoms related to compression of advanced tumors, including abdominal pain, abdominal masses, and jaundice. 7
At present, the diagnosis of this disease remains challenging, with pathological diagnosis through biopsy being the most reliable method. Imaging modalities, including multi-slice spiral computed tomography (CT) and magnetic resonance imaging (MRI), offer diagnostic value but often fail to distinguish PHNETs from other hepatic malignancies.8,9 Nevertheless, positron emission tomography/computed tomography (PET-CT) has high sensitivity and spatial fraction in detecting these tumors. 10 Surgery remains the most effective treatment for PHNETs, with a reported 5-year survival rate of 78%.11,12 In addition to surgery, alternative therapies such as transcatheter arterial chemoembolization (TACE), chemotherapy, and radiofrequency ablation (RFA) can prolong the patient’s mean survival when the tumor is unresectable.13–17
Despite advancements in diagnostic and therapeutic techniques, significant gaps remain in optimizing management strategies for PHNETs, particularly regarding high-quality evidence comparing the efficacy of different treatment approaches. Therefore, this review aims to systematically analyze existing literature and synthesize insights from population databases, including SEER-derived data, to comprehensively examine the clinical characteristics and prognostic factors of PHNETs. Furthermore, we will evaluate overall survival (OS) and disease-specific survival (DSS) across different treatment regimens, with particular focus on key variables such as tumor grade, to provide evidence-based references for clinical practice.
Methods
Based on the U.S. National Cancer Institute’s SEER database (1975-2019), we identified 446 patients with PHNETs from an initial cohort of 530 (see Figure 1). Demographic, clinical, and treatment data were analyzed. Survival outcomes, including OS and DSS, were evaluated using Kaplan-Meier method with log-rank testing, while a multivariate Cox proportional hazards model identified prognostic factors, reporting hazard ratios (HR) with 95% confidence intervals (CI). A P-value <0.05 was considered statistically significant for all two-sided tests, performed using IBM SPSS Statistics 25.0. The use of de-identified, public data exempted the study from IRB review and informed consent. Flowchart outlining the inclusion and exclusion criteria for primary hepatic neuroendocrine tumor (PHNET) cases from the SEER database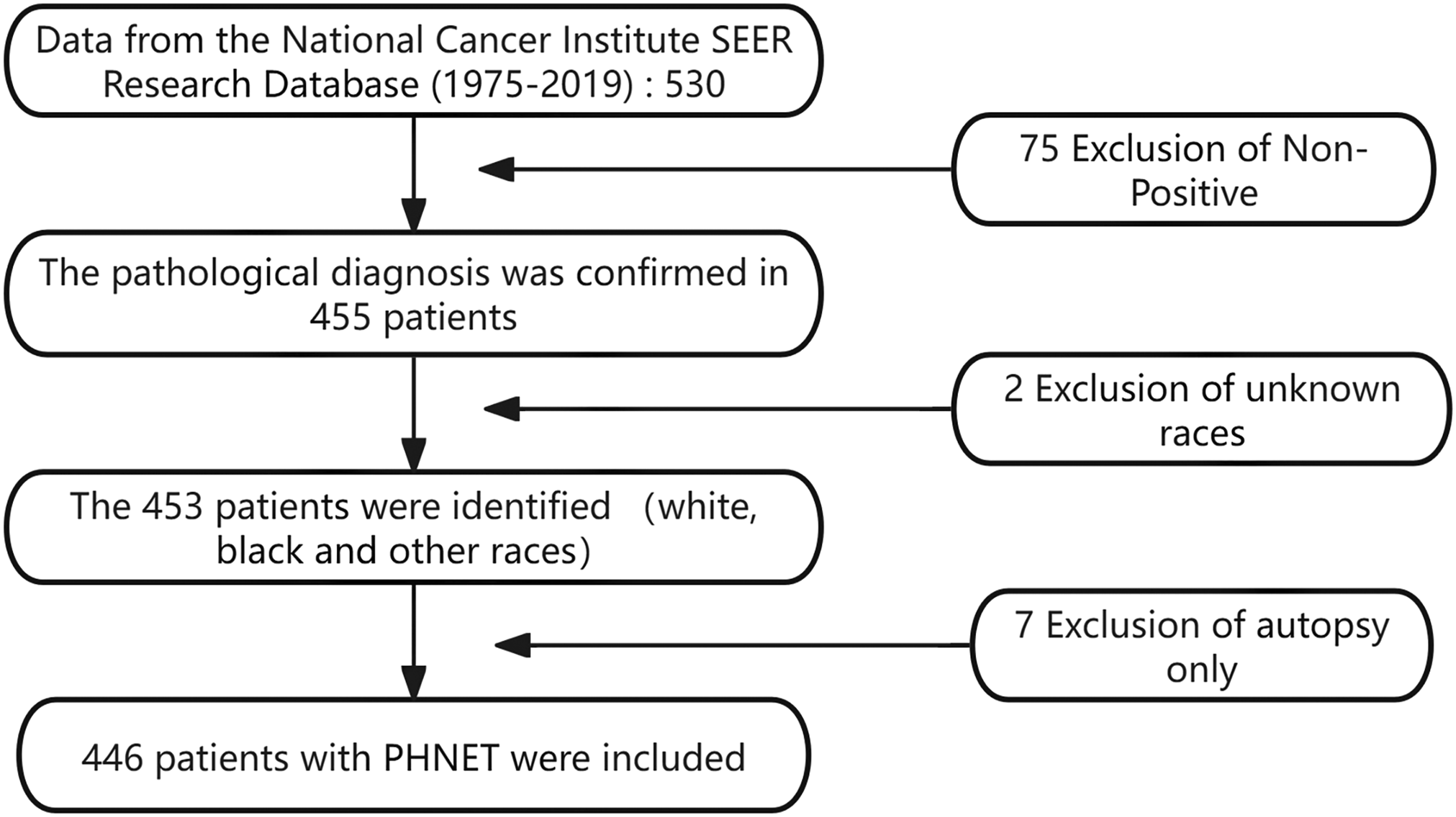
Results
Baseline Demographic and Clinical Characteristics of Patients Diagnosed With Primary Hepatic Neuroendocrine Tumors
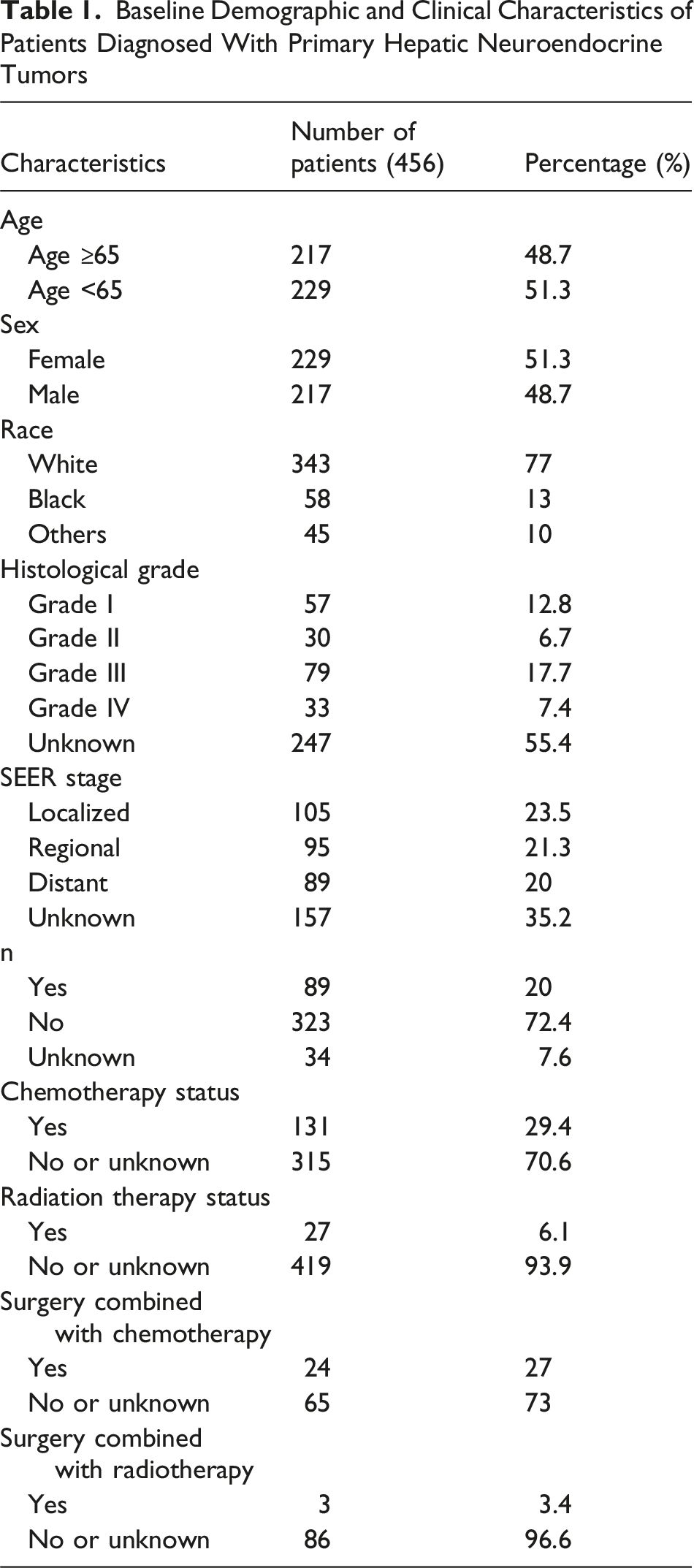
Regarding tumor grade, there were 57 patients with grade I, 30 with grade II, 79 with grade III, 33 with grade IV, and 247 with unknown tumor grades. Based on tumor stage in SEER, 105 patients had localized lesions, 95 had regional lesions, 89 had distant metastatic lesions, and 157 had unknown or blank lesions. A total of 89 patients underwent tumor-related surgery, while 323 did not, and surgical information was missing for 34 patients. Among those who underwent surgery, 24 received concurrent chemotherapy.
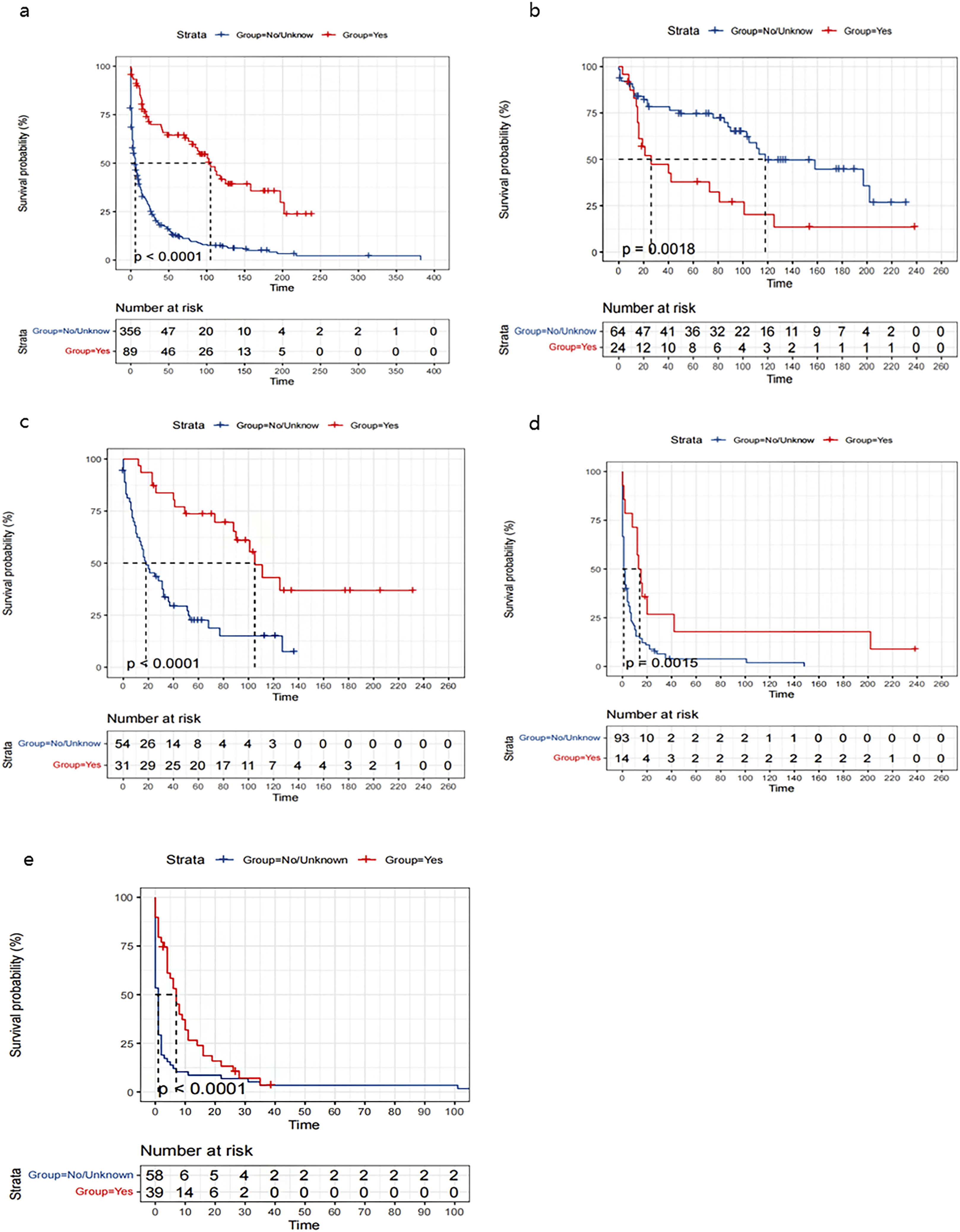
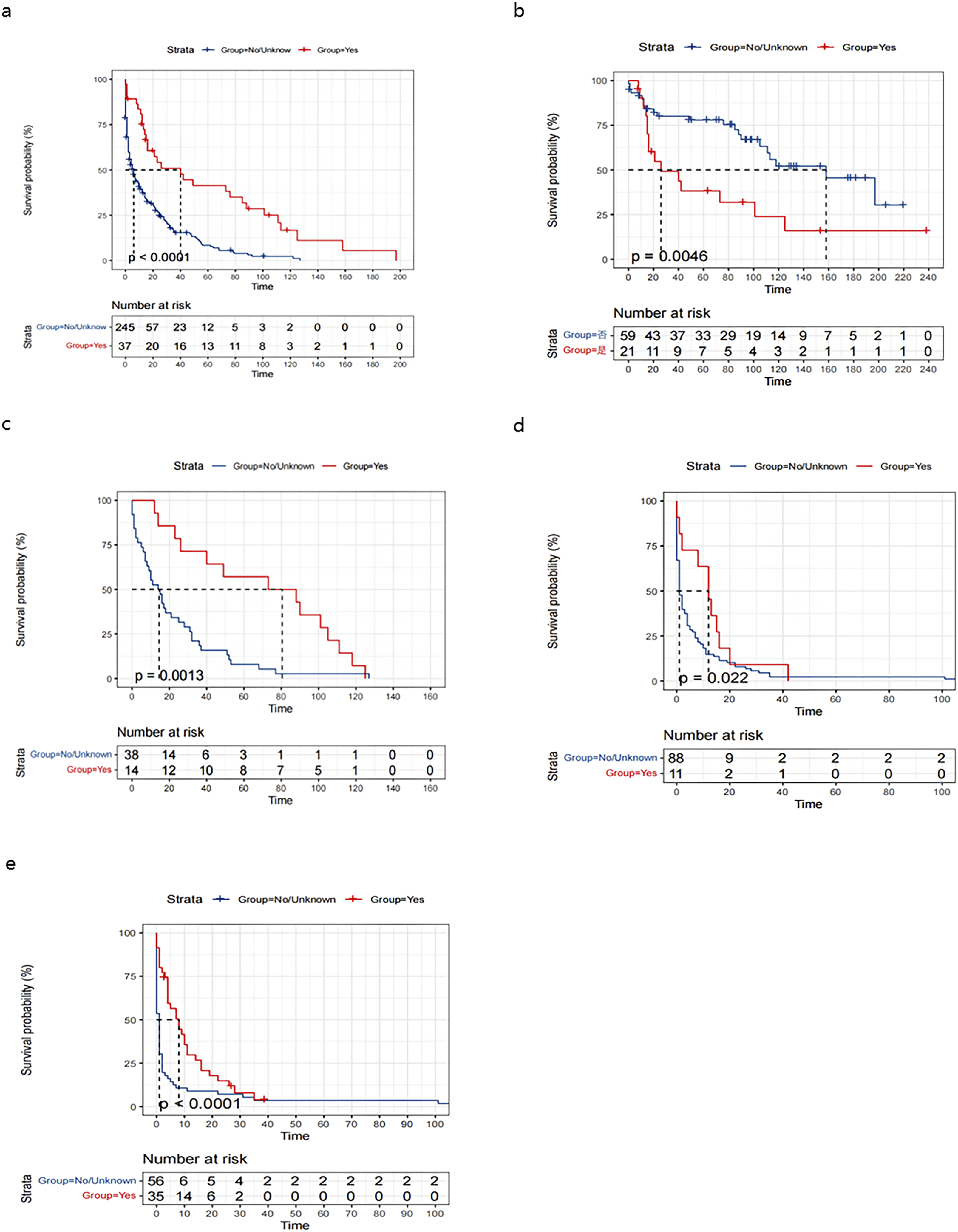
Multivariate analysis identified age ≥65 years (HR 1.56, 95% CI 1.24-1.93), higher tumor grade (G3: HR 2.94, 95% CI 1.93-4.48; G4: HR 3.04, 95% CI 1.83-5.07 vs G1), and no tumor-related surgery (HR 1.52, 95% CI 1.08-2.14) as independent predictors of poorer overall and disease-specific survival (all P < 0.0001, see Figure 2). Treatment modality analysis demonstrated superior survival with surgical intervention, particularly when combined with chemotherapy. In patients with high-grade tumors (G3 + G4), chemotherapy alone provided a significant survival benefit vs no treatment (see Figures 3 and 4). Multivariate Cox proportional hazards regression analysis for (A) overall survival (OS) (Figure 2(A)) and (B) disease-specific survival (DSS) (Figure 2(B)) Kaplan-Meier analysis of overall survival in patients with PHNET, stratified by: (A) tumor-directed surgery, (B) tumor-directed surgery combined with chemotherapy, (C) patients with G1 and G2 tumors receiving surgery, (D) patients with G3 and G4 tumors receiving surgery, and (E) patients with G3 and G4 tumors receiving chemotherapy alone Kaplan-Meier analysis of disease-specific survival in patients with PHNET, stratified by: (A) tumor-directed surgery, (B) tumor-directed surgery combined with chemotherapy, (C) patients with G1 and G2 tumors receiving surgery, (D) patients with G3 and G4 tumors receiving surgery, and (E) patients with G3 and G4 tumors receiving chemotherapy alone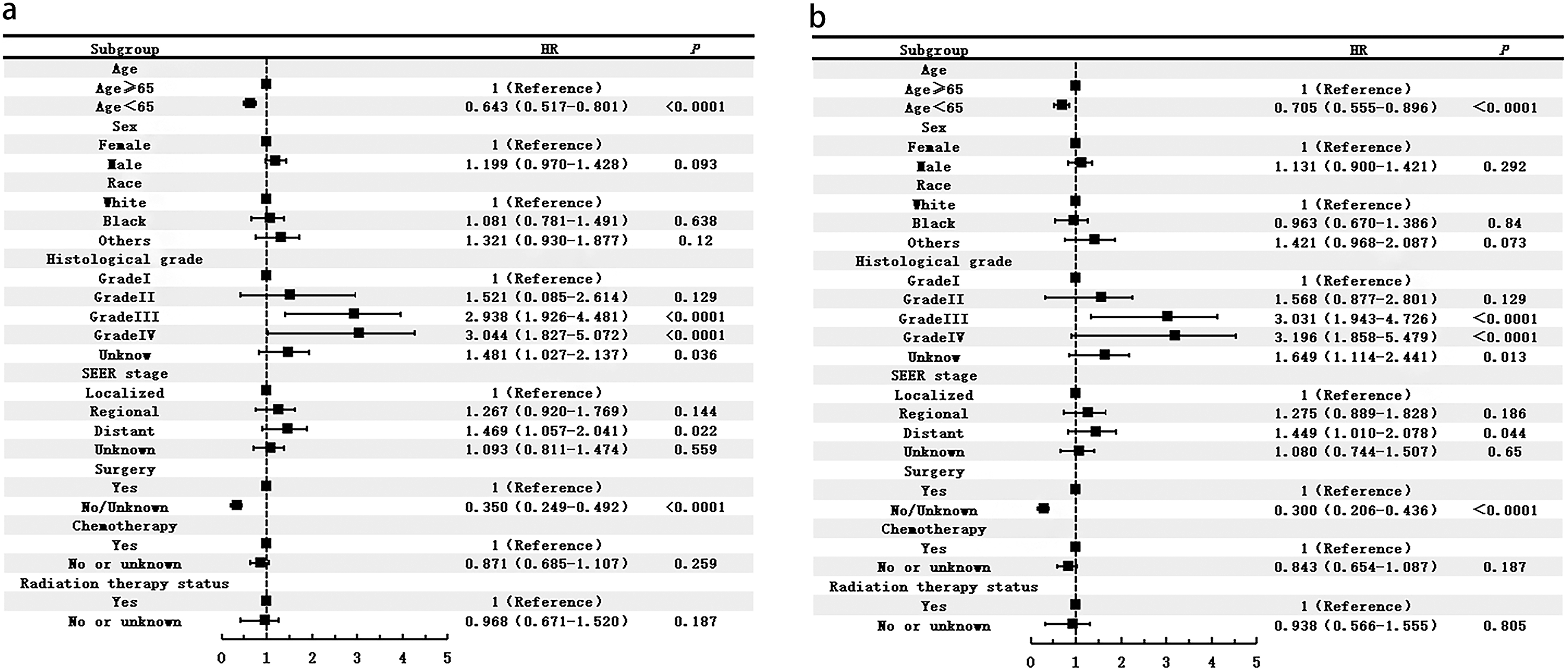
Discussion
The key remaining challenge in PHNET is not simply diagnosis but translating diagnosis into an operative plan. As summarized in Figure 5, we propose a stepwise decision-making framework for surgeons: first, confirm that the liver lesion represents a true primary rather than metastatic NET; second, define tumor biology by histologic grade; third, assess hepatic tumor burden, distribution, and technical resectability; and fourth, match these findings to the most appropriate operative or nonoperative strategy. This structure helps convert a descriptive review into a practical pathway for determining what operation to perform, in which patient, and why.12,18-20 Step 1: Confirm primary hepatic disease. Because the liver is the most common site of NET metastasis, the first surgical question is whether the lesion is truly primary.
19
Conventional imaging with ultrasound, CT, and MRI is useful for lesion detection but lacks specificity, and PHNET may mimic hepatocellular carcinoma or metastatic NET.21-25 Therefore, preoperative evaluation should systematically exclude an extrahepatic primary by combining cross-sectional imaging, somatostatin receptor PET-CT, targeted upper and lower gastrointestinal endoscopy, and biopsy with immunohistochemistry when needed.10,19,26,27 This is the critical gatekeeper step: if an extrahepatic primary is identified, the case should be managed as metastatic NET rather than PHNET, and routine curative-intent hepatectomy is generally not the correct first strategy. Step 2: Define tumor biology and operative intent by grade. Histopathological confirmation remains essential because grade directly affects prognosis and treatment intensity. In our analysis, higher grade independently predicted worse survival (G3: HR 2.94; G4: HR 3.04 vs G1), supporting grade as a core determinant of operative planning.
12
Well-differentiated low-grade tumors (G1/G2) are the best candidates for curative-intent liver resection when technically feasible, whereas high-grade tumors (G3/G4), especially those with rapid progression or extensive disease, more often require systemic therapy-first or multimodal treatment rather than upfront surgery alone.12,14,17 Neuroendocrine markers such as Syn, CgA, CD56, and NSE confirm neuroendocrine differentiation, while CDX2 or TTF-1 may suggest a non-hepatic primary and redirect treatment away from PHNET-specific surgery.26,27 Step 3: Assess tumor burden, distribution, and resectability. After confirming PHNET and defining grade, the next decision is whether the liver disease is localized and resectable, liver-confined but unresectable, or advanced and not amenable to meaningful cytoreduction. Solitary or limited tumors with an adequate future liver remnant should proceed to hepatectomy, with either anatomic or parenchymal-sparing resection selected according to lesion location and liver reserve.12,20 In contrast, multifocal bilobar disease, major vascular involvement, or inadequate remnant liver may preclude standard resection even in low-grade tumors. In these patients, the surgeon must distinguish between unresectable but liver-confined disease, where transplantation may be considered in carefully selected G1/G2 cases, and disseminated or biologically aggressive disease, where operative benefit is limited.13,20,28 Step 4: Match findings to treatment. This framework leads to 4 practical treatment pathways. First, patients with confirmed PHNET, G1/G2 biology, and localized resectable disease should undergo surgical resection, which remains the treatment most consistently associated with long-term survival.11,12,20 Second, highly selected patients with low-grade, unresectable but liver-confined disease may be considered for liver transplantation.13,20,28 Third, patients with multifocal unresectable disease, high hepatic tumor burden, or symptoms but without a clear resection strategy are better served by liver-directed therapies such as TACE or TAE for disease control and palliation.14,15 Fourth, patients with G3/G4 tumors, rapid progression, extrahepatic spread, or poor operative candidacy should be prioritized for systemic therapy and multidisciplinary management, including chemotherapy, somatostatin analogs, targeted therapy, or PRRT where appropriate.14,16,17 In this way, Figure 5 makes explicit the decision sequence that is otherwise only implied in the literature. Stepwise decision-making framework for PHNET diagnosis and management, from confirmation of primary disease to treatment selection based on grade, tumor extent, and resectability
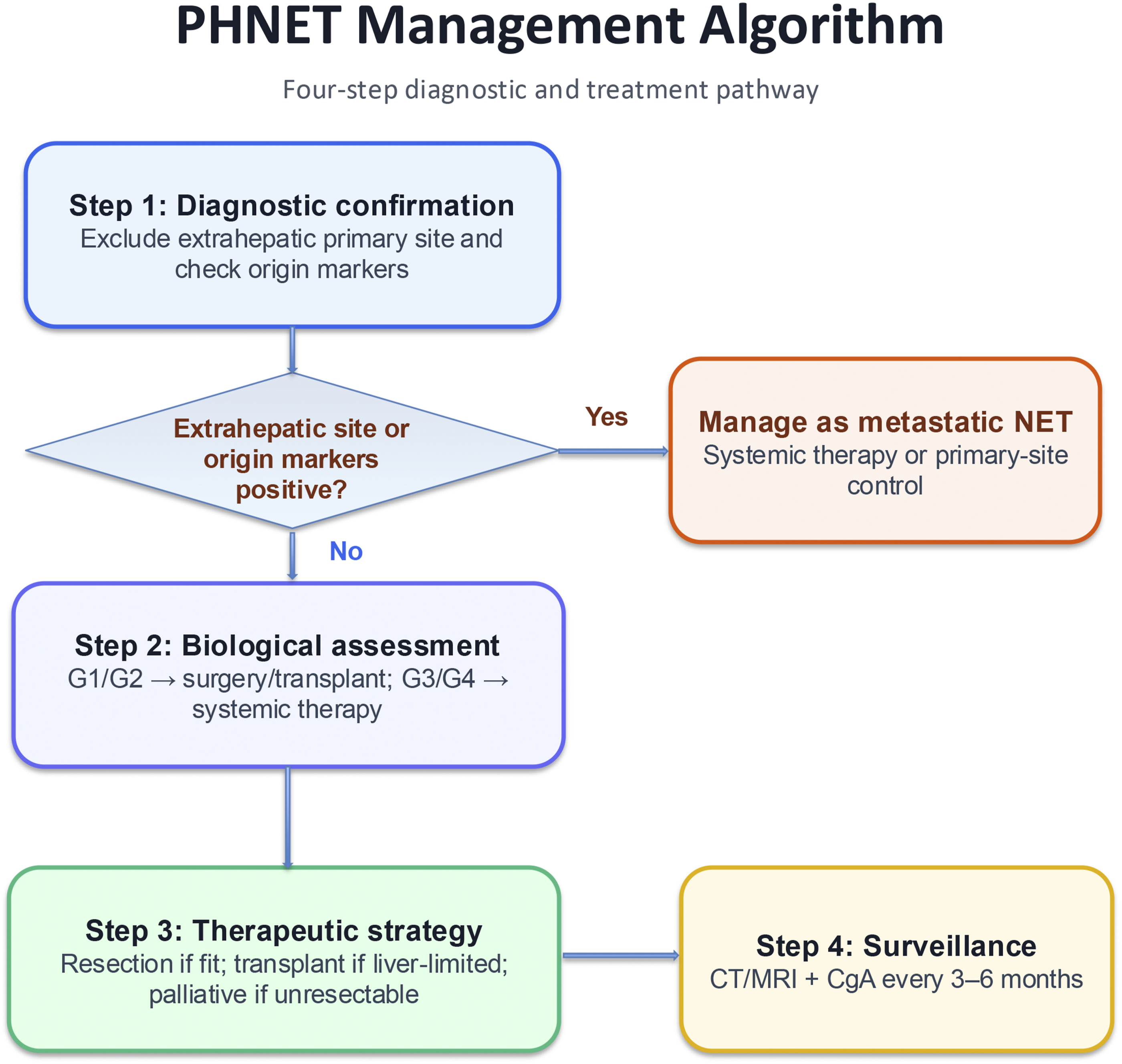
Post-treatment management should also follow the framework. Patients treated with resection or transplantation require structured surveillance with cross-sectional imaging and neuroendocrine markers, while those with high-grade tumors or uncertain primary origin warrant closer follow-up because of the higher risks of recurrence, progression, or later identification of an occult extrahepatic primary. In practice, this reinforces that PHNET management is not a one-time operation but an MDT-guided longitudinal strategy spanning diagnosis, operation selection, and surveillance.12,16
This analysis possesses several inherent limitations that warrant careful consideration. The extreme rarity of PHNETs inevitably results in treatment gaps and numerous undocumented or incompletely characterized cases within even large databases like SEER. Furthermore, the database structure does not capture granular details regarding surgical complications, postoperative course, or specific chemoradiotherapy regimens, preventing assessment of their impact on treatment outcomes and survival. These constraints may introduce bias and affect the precision of survival estimates presented herein.
Moving forward, future investigations would benefit from multi-institutional collaborative efforts that pool detailed clinical data, including standardized treatment protocols and comprehensive toxicity profiles. The establishment of an international PHNET registry would be particularly valuable in accumulating sufficient cases for meaningful subgroup analyses. Such prospective, systematically collected datasets will be essential for validating these findings and generating robust evidence to guide optimal management strategies for this rare disease entity.29,30
Conclusion
PHNETs are a rare disease. Factors such as age, ethnicity, sex, tumor grade, tumor stage, tumor-related surgical treatment, and the combination of surgery and chemotherapy have shown significant correlations with OS and DSS. Surgical resection remains the most reliable treatment for PHNETs, with improved outcomes observed when combined with chemotherapy. Treatment decisions, including surgery and chemotherapy, should be informed by the tumor grade to optimize patient prognosis.
Footnotes
Author Contributions
Conception and design of the research: Liang Yang and Yue Hu. Acquisition of data: Kaiyan Ling, Shuai Hua, and Lu Li. Analysis and interpretation of the data: Yue Hu, Yahui Liu, Guanbao Zhou, and Kaiyan Ling. Statistical analysis: Lu Li, Guanbao Zhou, Yahui Liu, and Shuai Hua. Obtaining financing: Liang Yang and Yue Hu. Writing of the manuscript: Lu Li and Kaiyan Ling. Critical revision of the manuscript for intellectual content: Liang Yang and Yue Hu. All authors read and approved the final draft.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Zhejiang Province Medicine and Health Science and Technology Program (2023KY1005). Zhejiang Province Medicine and Health Science and Technology Program (2024KY1512). Zhejiang Provincial Medicine and Health Science and Technology Program for Young Talents (2021RC21). Ningbo Natural Science Foundation (2021J310).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.