
Abstract
Background:
Given the link between cholesterol and activation of inflammation via interleukin 1β (IL-1β), we tested the effects of IL-1β inhibition on atherosclerotic calcification in mice. Patients with familial hypercholesterolemia develop extensive aortic calcification and calcific aortic stenosis. Although statins delay this process, low-density lipoprotein (LDL) cholesterol lowering alone is not enough to avert it. Data suggest that vascular inflammation initiated by hypercholesterolemia is followed by unchecked mineralization at sites of atherosclerotic plaques. The LDL-receptor (LDLR)-deficient (Ldlr−/−) and LDLR-attenuated Pcsk9(Tg) mice are available animal models for pharmacological testing.
Methods:
A mouse monoclonal antibody (mAb) against IL-1β or placebo was administered subcutaneously in Ldlr−/− and Pcsk9(Tg) models fed a Western diet. Drug level, anthropometric, lipid, and glucose profiles were determined. Expressions of proprotein convertase subtilisin/kexin type 9 (PCSK9), serum amyloid A1, and cytokine were measured by enzyme-linked immunosorbent assay. Aortic calcification was determined by microcomputerized tomography (micro-CT) and X-ray densitometry, and aortic flow velocity was assessed by ultrasound.
Results:
Circulating levels of IL-1β in Ldlr−/− mice were significantly greater (2-fold) than observed in Pcsk9(Tg) mice. Placebo- and mAb-treated mice did not differ in their growth, lipid, glucose profiles, and other cytokines. Calcifications were significantly diminished in mAb-treatment Ldlr−/− mice (a reduction of ∼75% by X-ray and ∼90% by micro-CT) and reduced insignificantly in mAb-treatment Pcsk9(Tg) mice, whereas aortic flow velocity was unchanged in both models.
Conclusions:
Herein, we demonstrate that aortic calcifications can be inhibited by an IL-1β mAb in LDLR-deficient mice. These results have a translational component to prevent vascular calcification in human and represent new evidence to rationalize targeting inflammation in cardiovascular disease.
Introduction
Patients with familial hypercholesterolemia (FH) as a result of mutations in the low-density lipoprotein receptor (LDLR) gene experience severe and extensive vascular calcification in an age-dependent fashion. 1 Computerized tomography (CT) scans of the thoracoabdominal aorta revealed that this phenotype involves the entire aorta by the second decade of life in homozygous FH. 1 In heterozygous individuals, a similar degree of vascular calcification is seen 20 years later. 2 In healthy, nonsmoking participants with no dyslipidemia or chronic metabolic disease, significant aortic calcifications are rarely seen before the seventh decade of life. Interestingly, all relevant clinical and laboratory parameters in relation to bone and calcium homeostasis are within reference values. 3 We recently recapitulated this phenotype in 2 mouse models and validated a micro-CT technique. 4 Using this method it was established that Ldlr knockout (KO) mice (Ldlr −/−) develop aortic calcification in a pattern similar to that seen in human. Furthermore, fewer calcifications were seen in Pcsk9(Tg) mice that are transgenic for proprotein convertase subtilisin/kexin type 9 (Pcsk9), 5 a protein known to promote the LDLR degradation. 6 Intriguingly, at a comparable level of serum cholesterol, an adult Ldlr−/− mouse fed a normal Chow-diet develops 70-fold increase in aortic calcification in comparison to a wild-type (WT) mouse fed a Western diet (WD). This led us to postulate a model in which vascular inflammation due to early hypercholesterolemia in FH is followed by dysregulated mineralization of subendothelial tissues. 7
The importance of vascular calcification in man is increasingly recognized in an aging population and in patients with FH. It is estimated that 30% of individuals older than 60 years of age have increased calcium deposits in major arteries. 8 The hemodynamic consequences of calcium buildup in the vasculature include a reduction in aortic and arterial compliance, and cardiovascular hemodynamic response becomes compromised. Clinically, the impact of vascular calcification contributes to arterial hypertension, aortic valve stenosis, limb ischemia, myocardial infarction, and congestive heart failure. 9 Furthermore, calcific aortic stenosis is the leading cause of aortic valve replacement in Western countries and the third leading cause of cardiovascular disease. 10 –13 It is estimated that in the Western world, approximately 3% of adults older than 75 years of age are affected by calcific aortic stenosis. 14
Unfortunately, at the present time no efficient therapy exists to stop or reverse vascular calcification. In addition, mixed results have been obtained with statins, especially the results from the Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) study were no benefit on the progression of aortic stenosis have been obtained. 15 Furthermore, statin combination therapy in patients with aortic stenosis was accused to augment cancer incidence, however, further analysis of the Study of Heart and Renal Protection (SHARP) and the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) has set the issue of cancer raised by SEAS to rest. 16
In the present study, we investigated whether inhibition of the inflammatory cytokine interleukin 1β (IL-1β) prevents vascular calcification. Interleukin 1β plays an essential role in triggering an inflammatory response to injury and mediating the calcification process via the actions of IL-1β on Wnt signaling. 7 Current data implicate cholesterol microcrystals as being the initiating factor in the release of IL-1β through the activation of the nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome. Inflammasomes are large, multimeric protein complexes that connect the metabolic stress sensing with proteolytic cleavage of pro-IL-1β into bioactive IL-1β. Inflammasomes are thus responsible for activation of inflammatory processes and have been shown to induce cell pyroptosis. 17 Stimulation of the IL-1 receptor on vascular endothelial cells leads to upregulation of inducible nitric oxide synthase, endothelin 1, chemokines/cytokines, adhesion molecules, endothelial and smooth muscle proliferation and macrophage activation, processes that contribute to endothelial dysfunction, and progression of atherosclerosis. 18 The targeted inactivation of IL-1β decreased plaque size in a mouse model of atherosclerosis (apoe −/− /IL-1β −/−) fed a high cholesterol diet. 19 In addition, lack of IL-1β decreased the severity of atherosclerosis in apoe −/− mice while prolonged treatment with IL-1β promotes arterial intimal thickening in the porcine coronary artery. 20 In addition, IL-1β has been implicated in atherothrombosis and plaque rupture in several preclinical and clinical studies. 20,21 Apparently, IL-1β function overlaps with tumor necrosis factor α (TNF-α) and several other cytokines. Furthermore, IL-1β is not embryonically lethal in mice. The IL-1β KO mouse has a normal phenotype, unless challenged with a pathogen. Furthermore, IL-1β inhibition does not modify plasma level of lipids and lipoproteins, thus making it a potential adjunct to current lipid lowering agents for the prevention and treatment of atherosclerotic vascular disease. Herein, we tested the hypothesis that inhibiting the early inflammatory process with a murine monoclonal antibody against IL-1β (01BSUR; Novartis, Basel, Switzerland), will lead to a marked reduction in vascular calcifications in mice.
Methods
Animal Protocol
Mice were fed a WD (Harlan Teklad # TD 88137) from weaning until euthanasia. The colony of Ldlr − / − mice (C57BL/6J background; stock 002529; n = 37) were purchased from The Jackson Laboratories (Bar Harbor, Maine). The Pcsk9(Tg) mice overexpressing PCSK9 (n = 31) were backcrossed to C57BL/6J for 10 generations. 5 Mice were kept in a pathogen-free facility under 12-hour light–dark cycles and fasted for 3 hours prior to euthanasia, and a total of 68 adult mice were used in this study with an equal ratio of males to females at the end of the study. All procedures were approved by the animal care committee of the Clinical Research Institute of Montreal. The study design is shown in Supplementary Figure 1. The protocol for genotyping and the sequence of used primers can be found in supplementary materials and Supplementary Table 1. The murine antimouse IL-1β monoclonal antibody (01BSUR, Novartis an IgG2a/κ monoclonal antibody) 22 was administered subcutaneously for 6 months. The half-life of the antibody in mice is approximately 14 days. The first and second doses were 10 mg/kg and subsequent injections were 7.5 mg/kg every other week for the remainder of the study. Conceptually, genetically modified mice deficient in LDLR or transgenic for Pcsk9, respectively, simulate homozygote and heterozygote FH phenotypes observed in humans. Treatment with either 01BSUR (treatment, +mAb) or NaCl 0.9% (placebo) was administered every 2 weeks after weaning and after the start of the WD. Thus, the following 4 groups were analyzed: (1) Ldlr −/− on placebo (n = 15), (2) Ldlr −/− on anti-IL-1β mAb (n = 15), (3) Pcsk9(Tg) on placebo (n = 13), and (4) Pcsk9(Tg) on anti-IL-1β mAb (n = 13).
Tissue Sampling and Plasma Analysis
Mice were analyzed at 4 and 6 months. Blood was collected at the induction of anesthesia by puncture of the left ventricle and then transferred immediately to heparin anticoagulated tubes on ice. Whole blood was used to measure glucose via a commercial glucometer (Bayer Diabetes Care, Leverkusen, Germany). Plasma was separated by centrifugation (20 minutes at 850g and 4°C) and kept at –20°C until assayed. Serum lipids (cholesterol, triglycerides, and high-density lipoprotein cholesterol after precipitation) were measured by an automated analyzer (COBAS INTEGRA 400; Roche Diagnostic, Indianapolis, USA) and LDL-C was derived. Mouse inflammatory cytokines were measured in 2 independent pools in each group by an expression enzyme-linked immunosorbent assay (ELISA) array (SABiosciences, Hilden, Germany; QIAGEN Company). Serum amyloid A1 (SAA1) and PCSK9 levels were measured by a quantitative ELISA kit (MyBioSource, San Diego, CA, USA and Cayman, Ann Arbor, MI, USA, respectively) according to the manufacturer’s recommendations. An in-house ELISA assay was developed by Novartis to measure the level of circulating IL-1β mAb (01BSUR), given that measuring IL-1β in injected mice with anti-IL-1β mAb would be misleading (interference with IL-1β assay). Thus, circulation levels of IL-1β mAb were determined by a competitive ELISA using highly purified goat anti-idiotypic antibodies raised against the Fab fragment of the mouse anti-IL-1β mAb. The ELISA is based on capture of a biotinylated anti-IL-1β mAb by plate-bound anti-idiotypic antibody and competition of this interaction by anti-IL-1β mAb present in the mouse serum. Serum concentrations of the antibody in mice were derived from standard curves generated with graded concentrations of the mouse anti-IL-1β mAb in mouse serum.
Aorta Isolation and Analysis
Under general anesthesia, mice were perfused and aorta fixed in situ as previously reported. 23 Fine dissection of the excised aorta was done under microscopic inspection, and each intact aorta was embedded in a single paraffin block for evaluation. For more details on surgical technique and histology protocol, see online Supplemental Material and Methods. Vascular calcification in humans can be detected on a standard X-ray or quantified more precisely on a CT scan. 2 –4,24 Thus, we applied a miniaturized version (Skyscan-1072, TomoNT version 3N.5, Belgium) to detect and quantify aortic calcification in mice with a resolution of 5 to 20 μm. A 3-dimensional reconstruction image and video (Supplementary Videos 1 and 2) were produced using commercial software to evaluate calcification. For details on X-ray and micro-CT settings and the assessments of aortic valve size using ultrasound and flow velocity, see online Supplemental Material and Methods and Supplementary Figure 2.
Statistical Analysis
Categorical variables are expressed as mean ± standard deviation. Commercially available software (GraphPad Prism software, La Jolla, California) was used for statistical analysis and preparing graphs. Analysis of variance between treatments groups (IL-1β mAb or placebo) was examined by Student t test. A P value <.05 was considered statistically significant. Simple random sampling was used to allocate animals to treatment groups. Sample size was based on our previous report using 6–month-old Ldlr −/− mice on WD 4 ; the mean aortic calcium score (AoCS) was 0.01% in WT mice fed a WD and 300-fold higher (3.0% ± 1.25%) in the Ldlr −/− mice. This large effect size reflects the genetic background of our mice. In the present study, we conservatively planned for a 50% reduction effect size between mice treated with the IL-1β mAb and placebo. Planning for an α value of .05 and power of 80% to detect this difference, we expected that the minimum number of mice per group would be 6. We analyzed ∼10 mice at the end of the 6-month treatment period by X-ray densitometry and reserved the micro-CT for confirmation using 6 mice (minimal sample size).
Results
Expression of IL-1β in Aortic Tissue of Ldlr − / − and Pcsk9(Tg) Mice
Advanced atherosclerotic lesions were prominent in the aorta of Ldlr −/− and Pcsk9(Tg) mice. Expression of IL-1β was examined by immunohistochemistry (IHC), and calcium was examined by Alizarin red staining in atherosclerotic plaques of Ldlr −/− and Pcsk9(Tg) mice both fed a WD for 6 and 12 months, respectively. As described in Supplemental Material and Methods, aortic tissues were stained with appropriate primary and secondary antibodies. Note the intense subendothelial staining for IL-1β and Alizarin red (Figure 1A and B), reflecting subendothelial inflammation and vascular calcification.
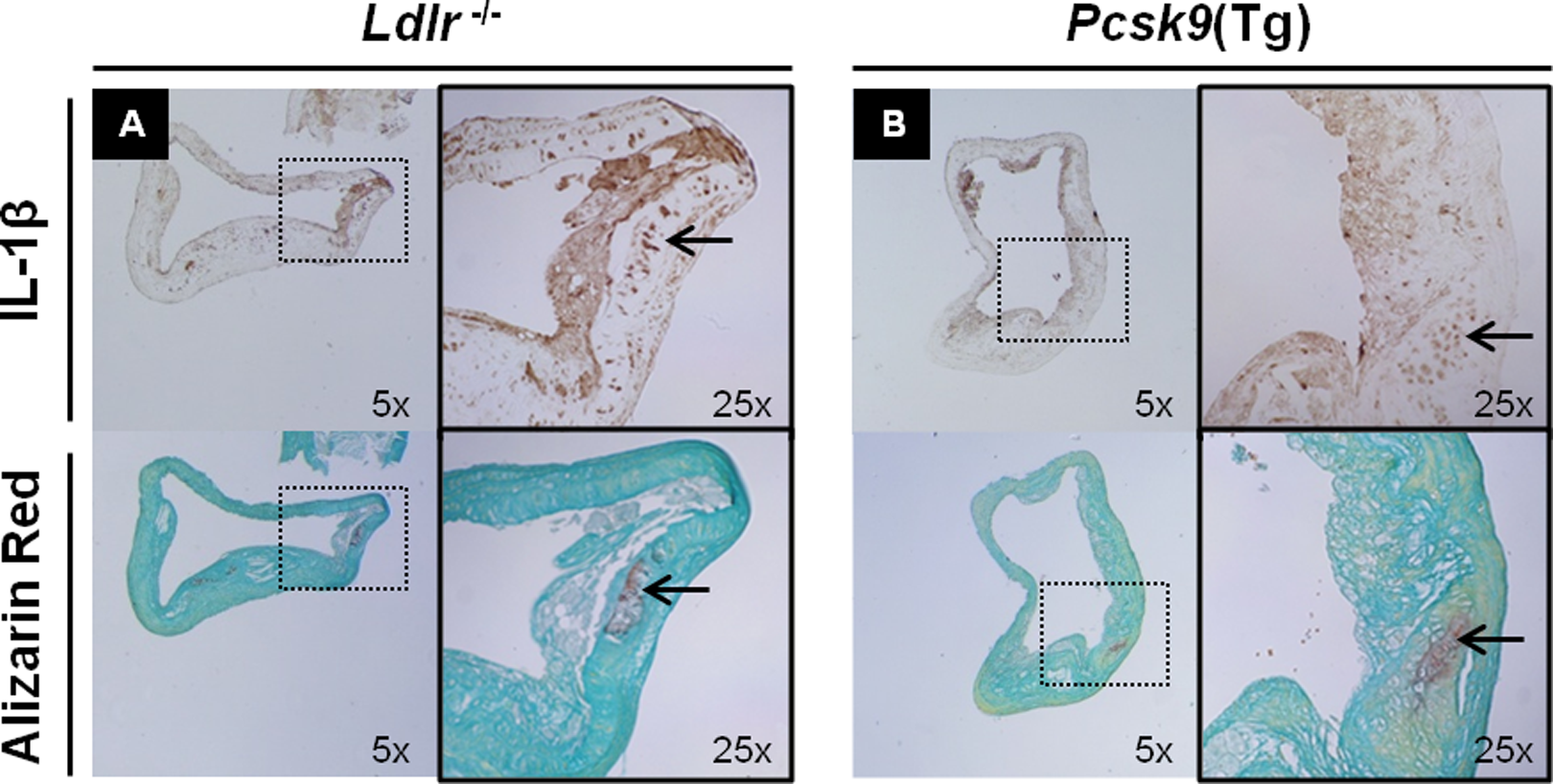
Tissue expression of IL-1β and Alizarin red in the aortic tissue of Ldlr − / − and Pcsk9(Tg) mice. A and B: Immunohistochemistry expression of IL-1β (top panel) and Alizarin red staining (bottom panel) in subendothelium area of ascending aorta in the Ldlr − / − (A) and Pcsk9(Tg) (B) mice fed a Western diet (WD) for 6 and 12 months, respectively. IL indicates interleukin.
Anti-IL-1β mAb treatment did not alter growth, glucose, and lipid profiles in comparison to placebo groups. Similar growth rates (Supplementary Figure 3) and organ weight profiles were seen after 6 months of WD in both arms. There were no significant differences between liver, kidneys, spleen sizes, and glucose levels between IL-1β mAb-treated and placebo-treated mice (Table 1). Lipid parameters were also not significantly changed between the 2 groups, including circulating PCSK9 levels between the Ldlr −/− mice groups (3287 ± 1194 vs 3793 ± 795 ng/mL; mAb-treatment vs placebo, respectively; P = .28) and between the Pcsk9(Tg) mice groups (35 273 ± 10 123 vs 29 899 ± 6591 ng/mL; mAb-treatment vs placebo, respectively; P = .44). As expected, Ldlr −/− mice had higher total cholesterol and triglyceride levels than Pcsk9(Tg) mice. Circulating plasma cholesterol levels were ∼2.5-fold higher in Ldlr − / − mice relative to Pcsk9(Tg) mice (Table 1), consistent with previous reports. 3
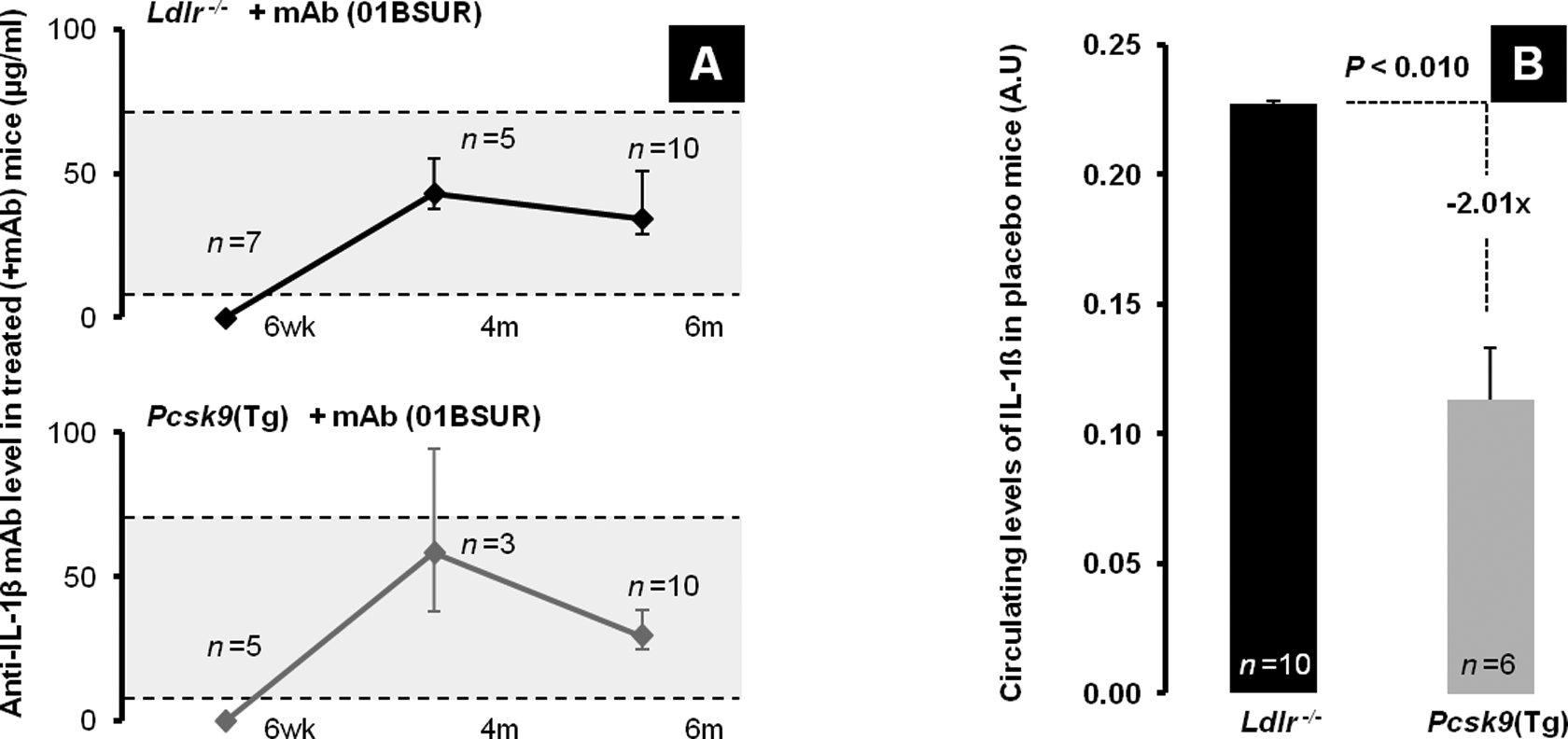
Drug levels and endogenous circulating levels of IL-1β in Ldlr − / − and Pcsk9(Tg) mice. A, Levels of anti-IL-1β monoclonal antibody (mAb; 01BSUR) at 6 weeks (before treatment), 4 and 6 months (after treatment). B, Circulating levels of IL-1β in Ldlr − / −and Pcsk9(Tg) mice on Western diet (WD) for 6 months. IL indicates interleukin.
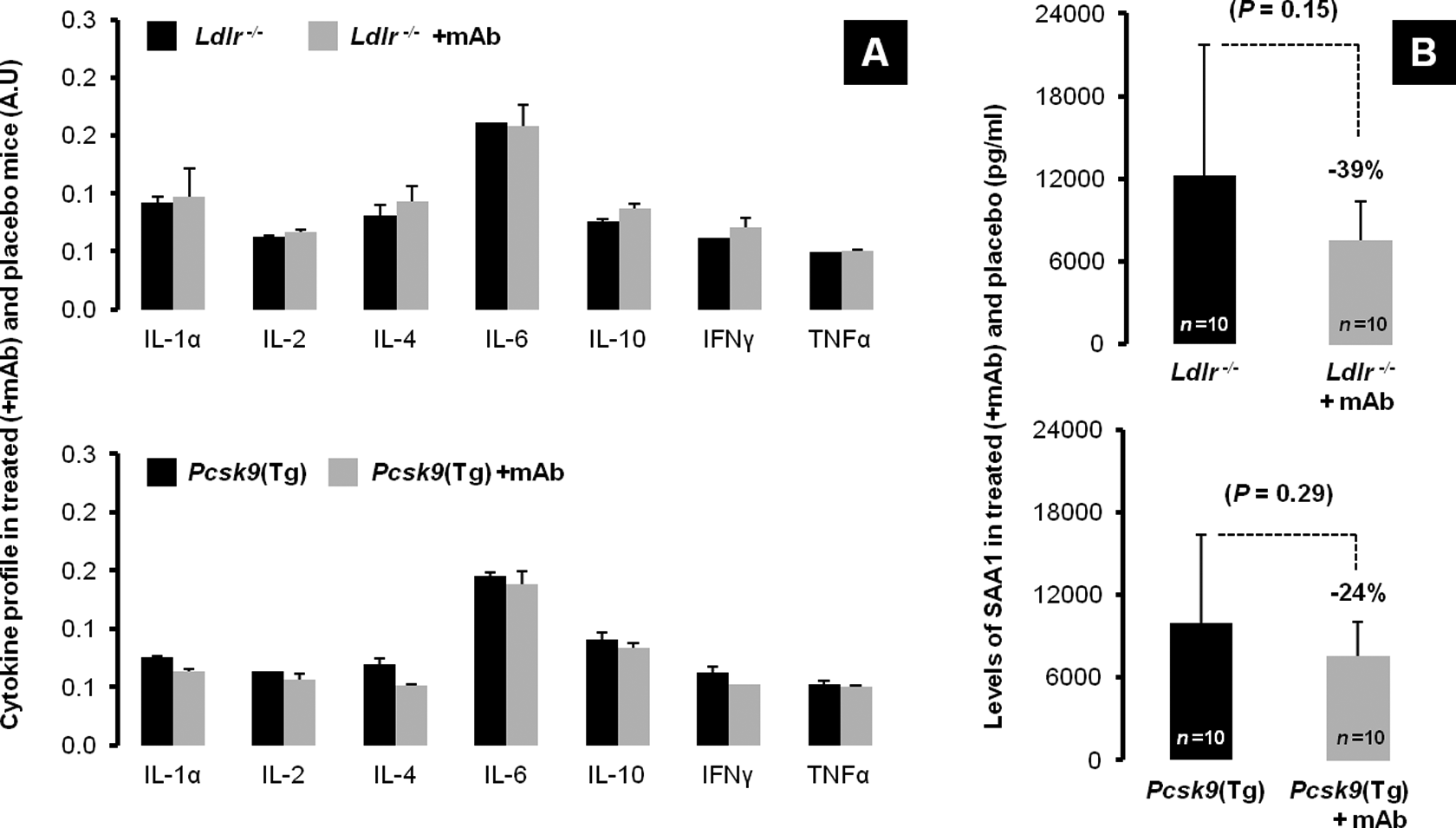
Cytokine levels and SAA1 in Ldlr − / − and Pcsk9(Tg) mice on different treatments. A, Placebo- and anti-IL-1β mAb-treated (+mAb) plasma levels of IL-1α, IL-2, IL-4, IL-6, IL-10, interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α) in both Ldlr − / −(top panel, all P values not significant) and in Pcsk9(Tg) mice at 6 months (bottom panel, all P values not significant). B, Plasma levels of SAA1 in both placebo- and mAb-treated (+ mAb) Ldlr − / −(top panel) and Pcsk9(Tg) mice (bottom panel). IL indicates interleukin; mAb, monoclonal antibody; SAA1, serum amyloid A1.
Attributes of Ldlr − / − and Pcsk9(Tg) Mice on Western Diet Assigned to Placebo or Treatment.a
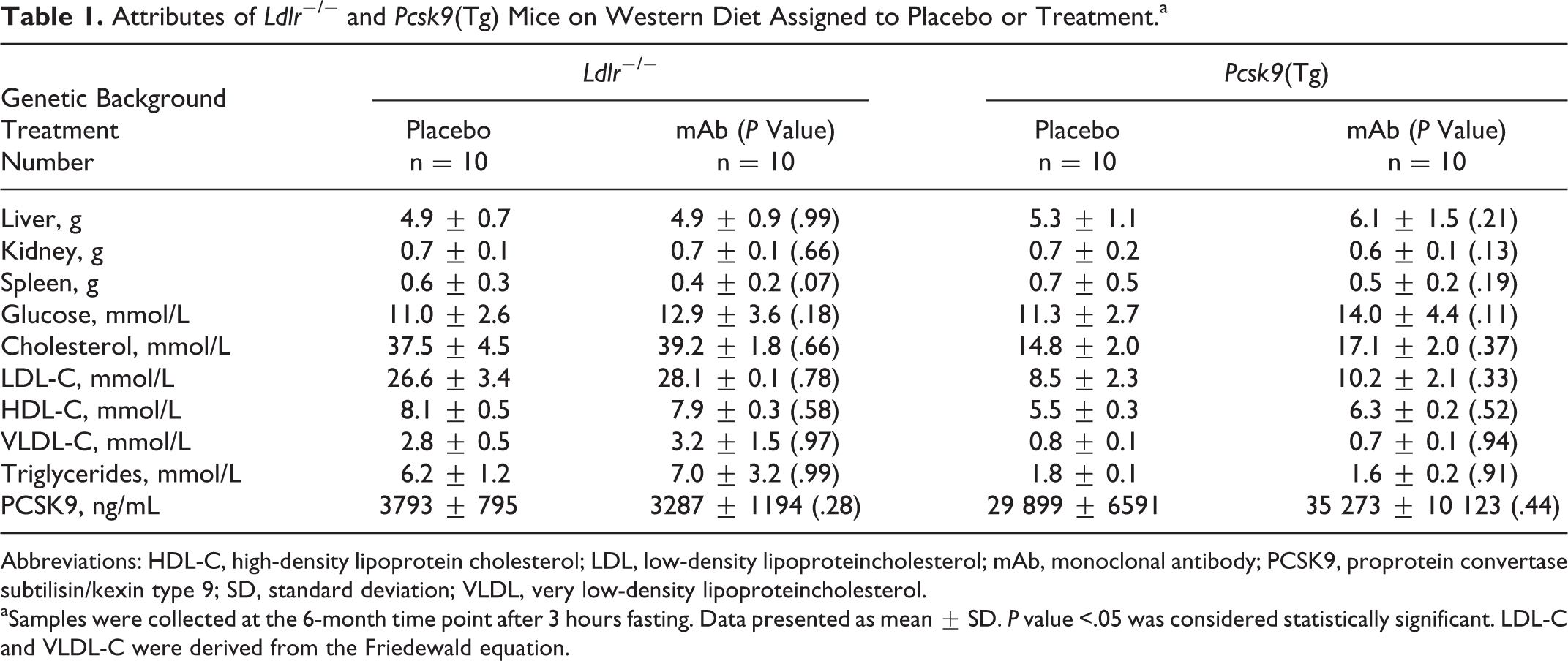
Abbreviations: HDL-C, high-density lipoprotein cholesterol; LDL, low-density lipoproteincholesterol; mAb, monoclonal antibody; PCSK9, proprotein convertase subtilisin/kexin type 9; SD, standard deviation; VLDL, very low-density lipoproteincholesterol.
aSamples were collected at the 6-month time point after 3 hours fasting. Data presented as mean ± SD. P value <.05 was considered statistically significant. LDL-C and VLDL-C were derived from the Friedewald equation.
Circulating Levels of Endogenous IL-1β Were 2-Fold Higher in Ldlr − / − Mice than Pcsk9(Tg) Mice
During the treatment phase, groups did not have major skin reactions or death related to repeated injections with anti-IL-1β mAb. To determine the presence of mAb treatment, levels of the anti-IL-1β mAb (bound and unbound 01BSUR) were determined by an ELISA at 6 weeks (prior to initiation of treatment), then at 4 and 6 months (during the trough window of treatment). As seen in Figure 2A, a measurable level of 01BSUR was maintained throughout the course of the treatment.
Previous reports have shown that endogenous IL-1β levels increase 2-fold in Ldlr − / − mice compared to WT, postlipopolysaccharide stimulation. 25 Inflammatory cytokines therefore were measured by an expression ELISA array (SABiosciences, QIAGEN Company). Endogenous levels of IL-1β were determined at 6 months in placebo-treated Ldlr − / − and Pcsk9(Tg) mice maintained on WD diet. Intriguingly, endogenous levels of IL-1β in Pcsk9(Tg) mice were half those observed in Ldlr − / − mice (P < .010; Figure 2B). Other than IL-1β, plasma levels of IL-1α, IL-2, IL-4, IL-6, IL-10, interferon γ, or TNF-α were not significantly different between Ldlr − / − and Pcsk9(Tg) mice on placebo nor between mAb treated and placebo treated on the same background (Figure 3A). Interestingly, the level of SAA1 between mAb-treated and placebo-treated groups was 39% lower in Ldlr − / − mice and 24% lower in Pcsk9(Tg) mice (but this did not reach statistical significance P = .15 and P = .29, respectively; Figure 3B). Considering the plasma levels of cholesterol, triglycerides, and IL-1β, the Ldlr − / − mice fed a WD show a more proatherogenic and proinflammatory profile than the Pcsk9(Tg) mice at 6 months of age.
Significant Reduction in Aortic Calcification in IL-1β mAb-Treated Ldlr − / − Mice
Ldlr
−
/
− mice fed a WD developed extensive atherosclerotic lesions as seen previously.4 Compared with placebo-treated Ldlr
−
/
− mice, IL-1β mAb-treated mice exhibited a marked decrease in aortic calcification, as measured by X-ray densitometry and by micro-CT AoCS (Figure 4A and B). As shown from X-ray densitometry, Ldlr
−
/
− mice on anti-IL-1β mAb showed a marked attenuation of vascular calcification by 4.1-fold (∼75%; 0.172 ± 0.167 in mAb vs 0.702 ± 0.625 in placebo, P = .013) and by 11-fold (∼90%) based on micro-CT scan quantification (0.27 ± 0.50 in mAb vs 2.95 ± 2.34 in placebo, P = .021; Figure 4C
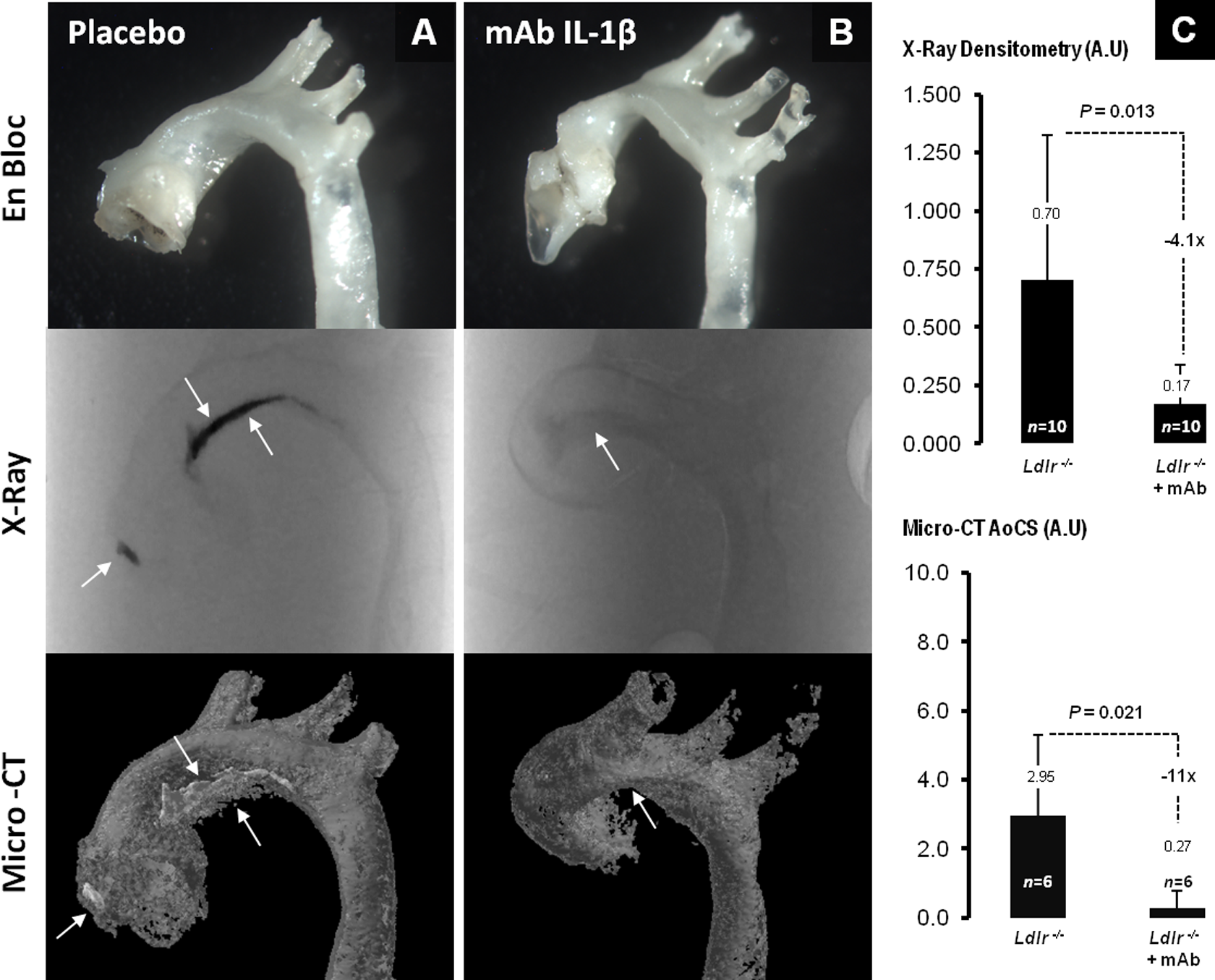
Significant reduction of aortic calcification in IL-1β mAb-treated Ldlr − / − mice. A, Aortas from placebo- and (B) mAb-treated Ldlr − / −mice. The top panel shows the thoracic aorta, removed en bloc, the middle panel shows the X-ray of the aorta, and the lower panel shows the 3-dimensional microcomputed tomography (micro-CT) reconstituted aortas; arrows point toward sites of calcification progression and resolution (C) shows the X-ray densitometry quantification (top) and aortic calcium score (AoCS; bottom) expressed in arbitrary unit (AU). IL indicates interleukin; mAb, monoclonal antibody.
As previously seen, the level of calcification in the Pcsk9(Tg) mice was lower and showed a later onset compared to Ldlr − / − mice. 4 In Pcsk9(Tg) mice, the effect of the IL-1β mAb treatment on parameters of aortic calcification was not significantly different between placebo- and mAb-treated groups at 6 months on WD (Figure 5A and B). The Pcsk9(Tg) mice on anti-IL-1β mAb showed a trend toward attenuation of vascular calcification by 55% as shown from X-ray densitometry (0.071 ± 0.089 in mAb vs 0.157 ± 0.135 in placebo, P = .090) and micro-CT scan quantification (0.0001 ± 0.00005 in mAb vs 0.0008 ± 0.0014 in placebo, P = .113; Figure 5C). Interestingly, Pcsk9(Tg) mice had levels of plasma lipids and IL-1β markedly lower than that of the Ldlr − / − mice (Table 1; Figure 2B).
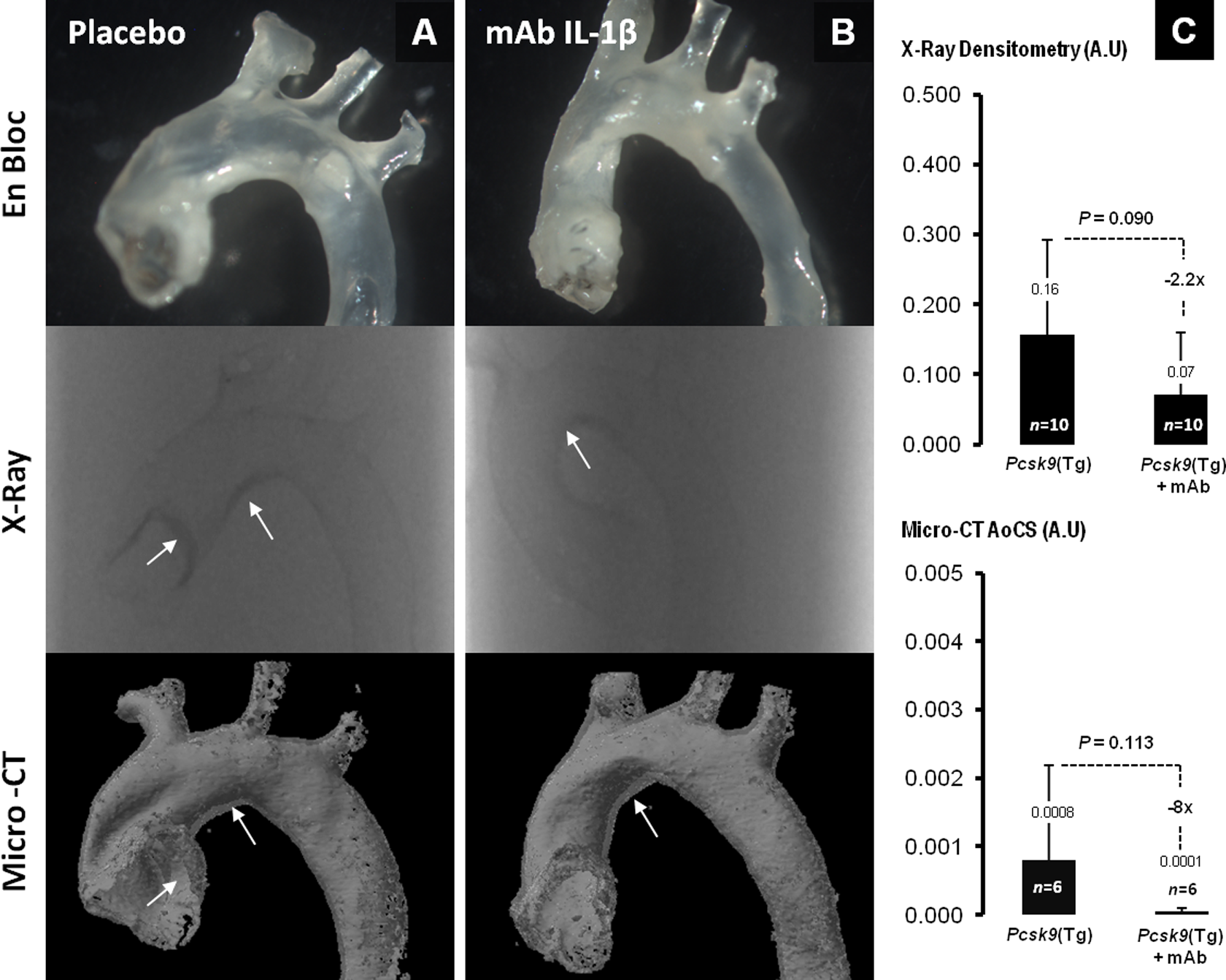
Attenuation of aortic calcification in IL-1β mAb-treated Pcsk9(Tg) mice. A, Aortas from placebo- and (B) mAb-treated Pcsk9(Tg) mice. The top panel shows the thoracic aorta, removed en bloc, the middle panel shows the X-ray of the aorta, and the lower panel shows the 3-dimensional microcomputed tomography (micro-CT) reconstituted aortas; arrows point toward sites of calcification progression and resolution (C) shows the X-ray densitometry quantification (top) and aortic calcium score (AoCS; bottom) expressed in arbitrary unit (AU). IL indicates interleukin; mAb, monoclonal antibody.
Expression of IL-1β by IHC and calcification by Alizarin red are shown in placebo and anti-IL-1β mAb-treated mice (Figure 6). Atherosclerosis develops in both Ldlr − / − and Pcsk9(Tg) mice after 6 months of WD, regardless of treatment status. However, advance aortic calcifications colocalize only with intense IL-1β staining in Ldlr − / − mice on placebo and are attenuated after the use of anti-IL-1β mAb (Figure 6A and B). In Pcsk9(Tg) mice, no advanced calcification lesions were seen at 6 months and thus anti-IL-1β mAb treatment have limited effect (Figure 6C and D).
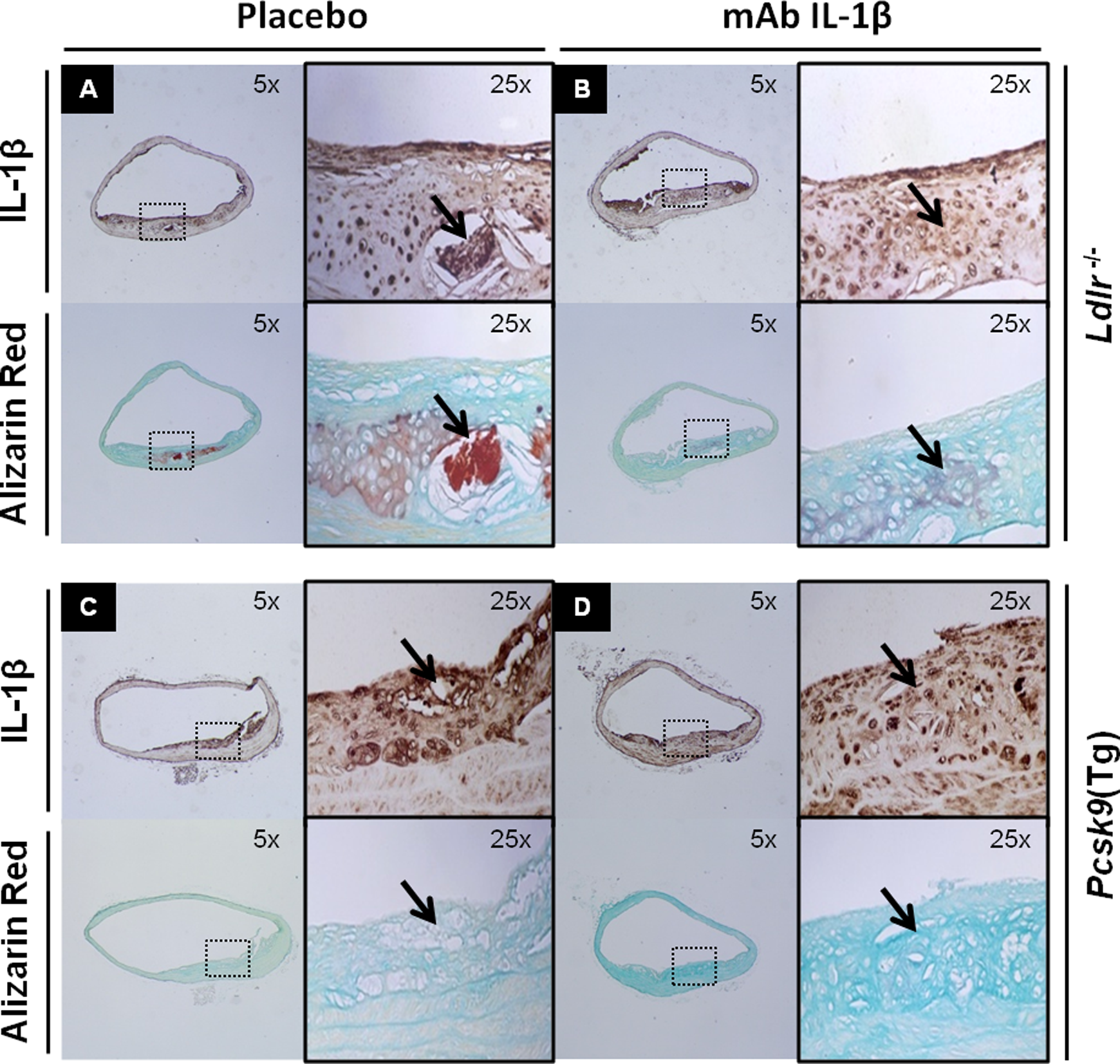
Expression of IL-1β by immunohistochemistry and calcification by Alizarin red in placebo- and mAb-treated mice. A and B, Ldlr − / − and (C and D) Pcsk9(Tg) mice after 6 months on Western diet (WD) develop subendothelial atherosclerotic lesions regardless of treatment status. Aortic calcifications colocalize with intense IL-1β staining in Ldlr − / − mice and calcification lesions were reduced by anti-IL-1β mAb treatment (A and B). In Pcsk9(Tg) mice, no advanced calcification lesions were seen at 6 months and thus anti-IL-1β mAb treatment had limited effects (C and D). IL indicates interleukin; mAb, monoclonal antibody.
Flow Velocities Were Unchanged Between Anti-IL-1β mAb and Placebo
Because humans with FH develop severe and premature aortic calcification, calcific aortic stenosis is often seen in patients with FH, especially in homozygous FH. We thus examined aortic flow velocity in Ldlr − / − and Pcsk9(Tg) mice by Doppler ultrasound. The difference in flow velocity across the valve in Ldlr − / − mice on anti-IL-1β mAb showed no significant difference from placebo (1.44 ± 0.34 in mAb vs 1.22 ± 0.30 in placebo, P = .15; Supplementary Figure 2).
Discussion
Familial hypercholesterolemia is a relatively common monogenic codominant Mendelian disorder and is caused predominantly by mutations in the LDLR gene, 26 coding for a cell surface glycoprotein that regulates plasma cholesterol via endocytosis of LDL particles. 27 A phenotype similar to FH can be caused by mutations in the LDLR ligand, apolipoprotein (apo) B gene (ApoB), and the regulatory protein produced by the PCSK9 gene. 28 At least in FH, the calcification process proceeds independent of cholesterol levels once subendothelial inflammation and atherosclerosis have occurred. 1 This supports a 2-hit hypothesis of vascular calcification, 7 where the first hit consists of intimal injury caused by lifelong hypercholesterolemia leading to accumulation of inflammatory cells, including monocytes/macrophages. The uptake of oxidized LDL by macrophages of the innate immune system is a critical step in the initiation of the atherosclerotic plaque and subendothelial inflammation. Recent data from Duewell et al 29 and Rajamäki et al 30 show that microcrystals of cholesterol are taken up by macrophages in the subendothelial layer of the arterial wall and activate the NLRP3 inflammasome, leading to the cleavage activation of pro-IL-1β and its release in plasma. The contemporary view of vascular calcification has been extensively reviewed. 7,8,15,31 –35
The second hit involves the calcification process by which calcium is deposited on matrix proteins in the intimal wall of genetically susceptible individuals. The involvement of inflammation in the calcification process has been shown previously. 18 –30 Importantly, IL-1β acts as a proximal amplification mechanism, causing the release of TNF-α and the activation of the Wnt pathway. 25 Furthermore, IL-1β has been implicated in the stimulation of alkaline phosphatase 36 and likely critical in the transdifferentiation of smooth muscle cells into an osteogenic phenotype. 21,37 Therefore, subsequent events leading to dysregulated mineralization have been postulated to involve a multistep pathway, which include subendothelial injury and release of IL-1β, activation of the Wnt canonical pathway of osteoblasts, and the decreased cross-talk between the LDLR and LRP5 cell surface receptors on osteoblasts. 7 This hypothesis establishes a link between atherosclerosis initiation, progression, and accelerated vascular calcification. Furthermore, this is also consistent with the results of several trials that have failed to show a benefit of statins in the progression of calcific aortic stenosis. 38 Indeed, intensive statin therapy in postmenopausal women for 1 year causes a marked LDL reduction than moderate therapy. However, this statin regiment did not result in delay of the progression of coronary calcification. 39
Indeed, C-reactive protein levels decreased within 1 week in patients treated with a fully human monoclonal antibody that neutralizes the bioactivity of human IL-1β. 40 By the time vascular calcification is detected, lipid-lowering therapy to prevent or reverse calcification appears to be of limited clinical benefit. Although statins may considerably slow down the process of atherosclerosis, they do not directly alter calcification. Conversely, elevated cholesterol seen in the WT mouse on a WD does not lead to aortic calcifications, in sharp contrast to the Ldlr − / − mouse model of aortic calcification. 4 Moreover, aged Ldlr − / − mice expressing ApoB100 developed extensive oxidative stress with aortic calcification and functional valvular disease. 41 Thus, our data indicated for the first time the potential role for IL-1β immune modulation in preventing vascular calcification.
In addition, to the presence of IL-1β in atherosclerotic aortic wall in Ldlr − / − mice (Figure 1), plasma levels of IL-1β were shown to be markedly higher in the Ldlr − / − mice compared to Pcsk9(Tg) mice (Figure 2B). This observation might explain why similar age Pcsk9(Tg) mice mount significant less calcification than Ldlr − / − mice and further confirm the atherogenicity of IL-1β. The Ldlr − / − mouse develop extensive aortic calcifications at 6 months on WD, while Pcsk9(Tg) mouse develop considerable calcifications only at 12 months of WD. 4 However, a milder calcification process was observed in the current report at 6 months augment by WD (Figure 5) and appropriate to the level of IL-1β.
It should be noted that our inhibitor is highly selective for IL-1β and does not cross-react with IL-1α. In addition, under our experimental conditions that does not involve pathogens, we would expect very little changes in cytokines reflecting activated immunity. Thus, downstream cytokine expression resulting in IL-1α and TNF-α release remains intact (Figure 3A). Although the lack of difference and change in other cytokines may come as a surprise, but in fact may support the selectivity of targeting IL-1β that does not disturb the immune homeostasis. Interestingly, the level of serum SAA1 (the major systemic marker of inflammation in mice) in Ldlr − / − and Pcsk9(Tg) mice was lower in treated mice in comparison to the placebo group, but this did not reach statistical significance probably due to the sample size of our study (Figure 3B). The PCSK9 level was also not suggestive of a direct relationship linking PCSK9 to atherosclerosis (Table 1).
Unfortunately, nuclear factor κB (NF-κB), a key player of inflammation, was not evaluated in the current study. The NF-κB pathway is divided into the canonical and noncanonical pathways. The canonical pathway is activated by TNF-α and IL-1β. Interestingly, IL-6 is a pleiotropic cytokine indicative also of inflammation and mineralization of the aortic valve in human. 42 However, the advantage of targeting IL-1β over IL-6 largely depends on the degree of immune inhibition necessary to inhibit subendothelium inflammation without developing immunodeficiency. Furthermore, it would be of importance to evaluate apoptosis mediated by ILs in animals treated with immune modulation, which may have detrimental consequences in the long run.
Nevertheless, after 6 months of treatment with an IL-1β mAb, Ldlr − / − mice showed a marked reduction in aortic calcification while Pcsk9(Tg) showed a nonstatistical trend toward reduced calcification. Perhaps due to lower basal hypercholesterolemia known in those mice compared to Ldlr − / − mice (Figure 5), since Pcsk9(Tg) represents heterozygous (partial absence of LDLR) and Ldlr − / − closely resembles homozygous (complete absence of LDLR).
The concept of immune modulation to prevent cardiovascular disease has been explored through studying animal models of IL-1β, IL-1 receptor deficiency, and IL-1 receptor antagonist deficiency. 43 –48 However, our study is novel in that it highlights vascular calcification as the primary end point of cardiovascular disease. Interestingly, human polymorphisms have been identified in the IL-1β gene. Some of these polymorphisms are important when translating finding from our study and therefore antibody binding site on the IL-1β should be characterized or analyzed in silico to determine whether particular polymorphisms will negatively impact the binding affinity of the IL-1β antibody.
In conclusion, as a proof of concept, the murine IL-1β monoclonal antibody (01BSUR) represents a potential therapy to prevent vascular calcifications in mice. Interestingly, a randomized phase III clinical trial is currently underway to investigate the effect of subcutaneous injections of a fully humanized antibody directed against IL1β, canakinumab, 49 in patients with stable postmyocardial infarction showing elevated levels of the inflammatory biomarker C-reactive protein. The incidence of FH is often quoted at 1:500 in the general population and it is much higher (1:80) in some communities with a founder effect. These patients are at potential risk of premature aortic calcification and late aortic complications that are associated with impaired functional status and survival. A translational component of this study therefore may apply to the overall treatment of patients with homozygous and severe heterozygous FH. Individuals with gain of function mutation in PCSK9 gene that promotes the LDLR degradation can similarly benefit from this approach.
Footnotes
Acknowledgments
We thank Suzie Riverin, Marie-Claude Lavallée, and Jade Dessureault (Animal facility) for excellent animal care; Dominique Lauzier (Histology facility), Dominic Filion (Microscopy facility), May Faraj, Dany Gauthier, and Isabelle Ruel for technical assistance; and Manon Laprise (Ultrasound platform), Yongjun Xiao (mico-CT platform), and Amr Awan (Database Management).
Authors’ Note
No funds were received from Novartis pharmaceutical, other than supplying the study team with IL-1β monoclonal antibody (01BSUR) and were not involved in manuscript drafting. HG is the study collaborator and the scientist at Novartis Institutes for Biomedical Research. Dr HG holds no ownership of stock or other equity position for (01BSUR) used in the study nor received honoraria or consulting fees for his collaboration.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Seraj Kaki Chair for the study of genetic polymorphisms in cardiovascular disease and diabetes, grant # MBK/03/434 (to ZA), King Abdulaziz University, Jeddah, Saudi Arabia, and the Canadian Institutes of Health (CIHR) CIHR Team grants CTP 82946 on Convertases in Cardiovascular Diseases, CIHR MOP 93792, and a Canada Chair 216684 (to NGS), Ottawa, Canada.