
Abstract
Numerous studies have shown that a low level of high-density lipoprotein cholesterol (HDL-C) is an independent biomarker of cardiovascular disease. High-density lipoprotein (HDL) is considered to be a protective factor for atherosclerosis (AS). Therefore, raising HDL-C has been widely recognized as a promising strategy to treat atherosclerotic cardiovascular diseases (ASCVD). However, several studies have found that increasing HDL-C levels does not necessarily reduce the risk of ASCVD. HDL particles are highly heterogeneous in structure, composition, and biological function. Moreover, HDL particles from atherosclerotic patients exhibit impaired anti-atherogenic functions and these dysfunctional HDL particles might even promote ASCVD. This makes it uncertain that HDL-raising therapy will prevent and treat ASCVD. It is necessary to comprehensively analyze the structure and function of HDL subfractions. We review current advances related to HDL subfractions remodeling and highlight how current lipid-modifying drugs such as niacin, statins, fibrates, and cholesteryl ester transfer protein inhibitors regulate cholesterol concentration of HDL and specific HDL subfractions.
Keywords
Introduction
Atherosclerosis (AS), a chronic and progressive vascular disease, is the leading cause of cardiovascular (CV) morbidity and mortality worldwide. AS is characterized by excessive cholesterol deposition in the arterial intima, which leads to atherosclerotic plaque and subsequent thrombus formation, increasing the risk of CV disease (CVD). 1
The inverse relationship between high-density lipoprotein cholesterol (HDL-C) level and the risk of clinical events resulting from AS is consistently recognized across numerous clinical and epidemiological studies.2,3 Therefore, targeting high-density lipoprotein (HDL) for the treatment of atherosclerotic CVD (ASCVD) presents an attractive strategy. Nevertheless, the assumption that increasing HDL-C levels would be beneficial to ASCVD has been questioned. Treatment with elevated HDL-C did not improve CV outcomes in the majority of clinical trials. 4 Some atherosclerotic CV patients have normal, or even high levels of HDL-C. Intriguingly, some scientists proposed that CV mortality may increase at low and/or even extremely high HDL-C levels, showing a U-shaped curve or a J-shaped relationship.5–8 The inconsistent relationship between HDL-C levels and CV risk may be attributed to several explanations. 9 First, a simple assessment of HDL-C levels might not be sufficient. The quantity of HDL-C provides limited information, while the property of HDL particles (HDLs) may provide more information regarding their functionality. Second, genetic factors could be another potential reason for different outcomes. Mutations in the gene encoding cholesteryl ester transfer protein (CETP) resulted in elevated HDL-C levels, but are inconsistent with the relationship with CVD. 10 Until now, almost all CETP inhibitors have been declared to fail to reduce the occurrence of CVD, despite the significantly elevated plasma HDL-C levels. Perhaps this may be attributed to the elevated HDL simply being large or even dysfunctional HDL.11–13 Therefore, some clinical trials using CETP inhibitors to reduce CV events did not achieve the expected results.
A topic of considerable interest is whether HDL subfractions are more informative in assessing the functionality of HDL. Numerous studies have demonstrated that the large and small HDL subfractions may differ in their ability to protect against ASCVD.14,15 Much remains unknown about the anti-atherosclerotic properties of HDL subfractions. This review mainly focuses on the dynamic remodeling and functional heterogeneity of HDL subfractions, which may provide critical information for the discovery of novel biomarkers and new pharmacological targets for ASCVD.
Composition, Heterogeneity, and Functionality of HDL
Basic Composition and Structure of HDL
HDLs are a class of lipoprotein populations that are small, dense, lipid-poor, and protein-rich, with a mean size of 7–12 nm and a density of 1.063–1.21 g/mL. 16 HDL mainly consists of a hydrophobic core of non-polar lipids, primarily triglycerides (TG), and cholesterol esters (CE), and a hydrophilic outer layer containing phospholipids (PL), apolipoproteins and free cholesterol (FC). 16 The further structural analysis confirmed the compositional complexity of HDL, which carries >80 different proteins, over 200 lipid species, as well as various microRNAs. 17 The protein components of HDL are exceedingly diverse, including structural apolipoproteins, enzymes, co-factors for enzymes, and many other proteins. 18 Among them, apolipoproteins and enzymes are generally recognized as crucial protein components. In addition to the protein constituent, approximately half of the total HDL mass is accounted by lipid components. 19 The majority of the HDL lipidome is composed of phospholipids, primarily phosphatidylcholine and sphingolipid species, which are dispersed on the surface of HDLs and form a monolayer of lipid molecules that stabilize the HDL structure. 20 Unesterified (free) cholesterol are located in the surface lipid monolayer of HDLs and regulate its fluidity. 14 Perhaps due to the high affinity of FC for sphingolipids, which tend to preferentially associate with large, light HDL. 19 Additionally, HDL also contains functional microRNAs, which suggests HDL can involve in cell-to-cell communication by delivering microRNAs. 21
Heterogeneity of HDL
Classification of HDL subfractions based on their physic-chemical properties. a

aHDL: high-density lipoprotein; LpA-I: containing apolipoproteins A-I (ApoA-I) but not apolipoproteins A-II (ApoA-II); LpA-I: A-II: containing ApoA-I and ApoA-II.
Functionality of HDL
HDL is thought to be a preventive and protective factor for ASCVD through its involvement in the reverse cholesterol transport (RCT) process. RCT refers to a physiological process by which excess cholesterol in peripheral cells is transported through HDL to the liver, where it can be directly excreted into the bile or metabolized into bile acids/salts. It is widely considered the major mechanism responsible for HDL-mediated atheroprotection. Other properties which underlie the atheroprotective effects of HDL include anti-oxidant, anti-inflammatory, anti-thrombotic, and anti-infective, as well as cytoprotective and vasodilatory effects. 24 Not only that, scientists have found that HDL can influence glucose metabolism, increase plasma insulin levels and lower glucose concentrations in type 2 diabetes mellitus. 25 In addition, HDL reduced breast cancer metastasis by inhibiting the adhesion of breast cancer cells to endothelial cells and reduced cancer cell growth by inhibiting tumor angiogenesis. 26 However, several risk factors such as disorders of lipid metabolism, inflammatory response, oxidative stress, and immune damage have been found to cause the conversion of functional HDL to dysfunctional HDL. 27
HDL Biosynthesis, Remodeling, and Metabolism
The biosynthesis, remodeling, and metabolism of plasma HDLs are a series of complex processes. Many critical proteins and enzymes have been discovered to regulate the levels, composition and structure of HDLs. The impact of these main proteins and enzymes involved in these processes was summarized (Figure 1 and Table 2). Major pathways for the biosynthesis, remodeling and metabolism of HDL. Apolipoprotein A-I (ApoA-I) secreted by the liver and intestine accepts peripheral cellular phospholipids (PL) and free cholesterol (FC) via ATP-binding cassette transporter A1 (ABCA1) transporter to form small, discoid preβ-HDL. Subsequently, lecithin/cholesterol acyltransferase (LCAT) mediates cholesterol esterification and produces small HDL3 and large HDL2, which further accept PL and FC transported by scavenger receptor BI (SR-BI) and ATP-binding cassette transporter G1 (ABCG1). Moreover, small and large spherical HDL undergo further remodeling by hepatic lipase (HL), endothelial lipase (EL), lipoprotein lipase (LPL), phospholipid transfer protein (PLTP), and cholesteryl ester transfer protein (CETP). The cholesteryl esters (CEs) in HDL are metabolized primarily in the liver, either by selective uptake via SR-BI or CETP-mediated transfer to triglycerides-rich lipoproteins (TGRLs) and then is cleared indirectly by the low-density lipoprotein receptor (LDLR). The black line represents the biosynthesis process of HDL, the red line represents the remodeling process of HDL, and the orange line represents the metabolism process of HDL. Major proteins involved in HDL biosynthesis, remodeling, and metabolism.
a
aHDL: high-density lipoprotein; HDLs: HDL particles; HDL-CEs: HDL-cholesteryl esters; ApoA-I: apolipoprotein A-I; ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; LCAT: lecithin/cholesterol acyltransferase; HL: hepatic lipase; EL: endothelial lipase; LPL: lipoprotein lipase; PLTP: phospholipid transfer protein; CETP: cholesteryl ester transfer protein; SR-BI: scavenger receptor BI; LDLR: low-density lipoprotein receptor; LDL: low-density lipoprotein; PL: phospholipids; FC: free cholesterol; TG: triglyceride; TGRLs: triglycerides-rich lipoproteins.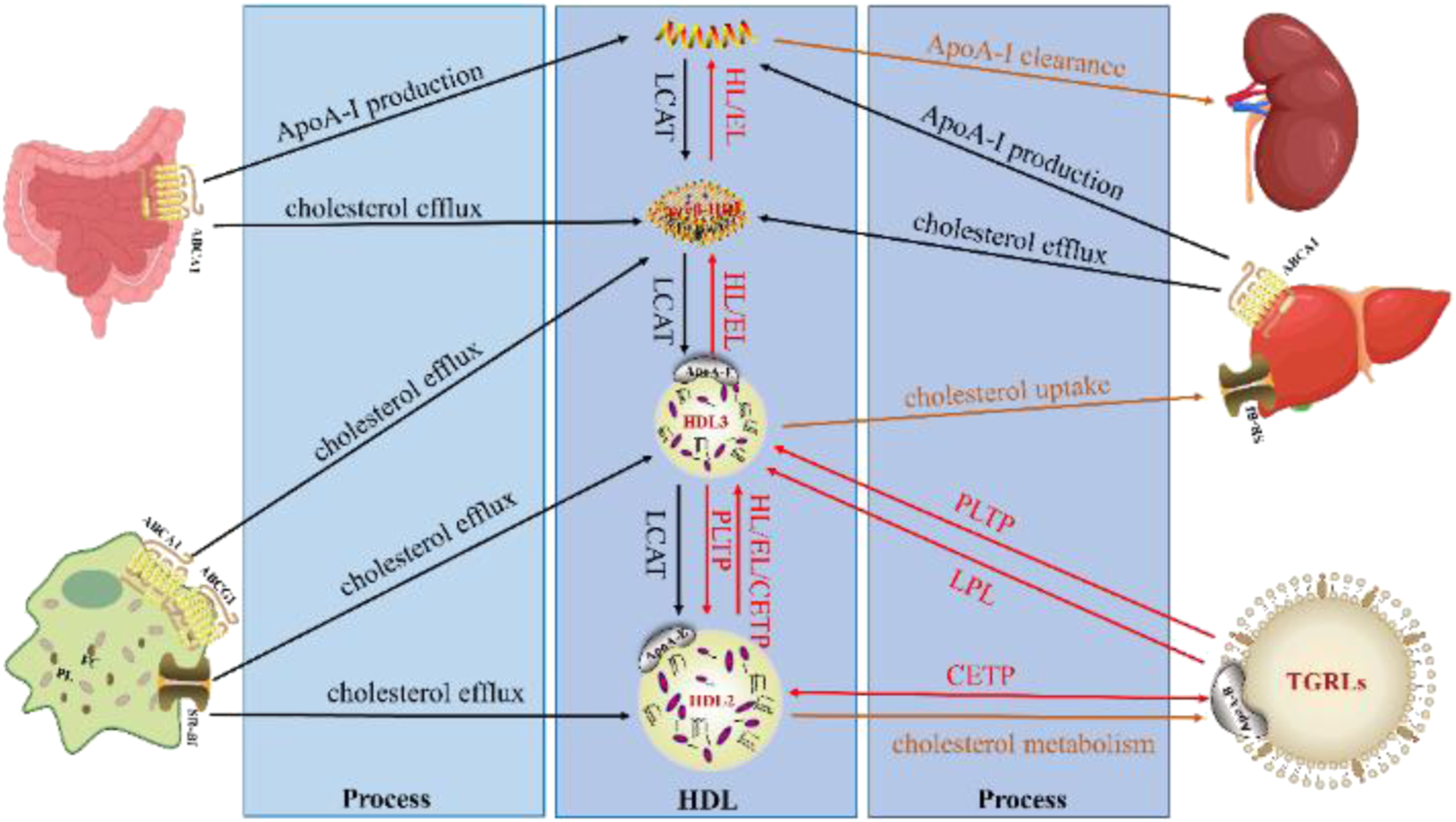

Biosynthesis of HDL
Plasma HDLs are synthesized through an orderly process. Lipid-poor ApoA-I secreted by the liver and intestine, 35 initially accepts peripheral cellular PL and FC transported by the cell surface ATP binding cassette A1 (ABCA1) to form nascent discoid HDL, also known as preβ-HDL.36,37 Subsequently, the cell surface scavenger receptor class B type I (SR-BI) and the ATP binding cassette G1 (ABCG1) can further mediate the lipidation of preβ-HDL to form larger and spherical HDL particles (HDL3). 38 FC in discoid and spherical HDLs is then esterified to CE by the enzyme lecithin/cholesterol acyltransferase (LCAT) and migrates into the core of the HDLs, leading to the formation of mature, spherical, α-migrating HDL particles (HDL2). 1 These HDLs synthesized in plasma undergo constant remodeling as they transport cholesterol between peripheral cells and other lipoproteins, leading to the mutual conversion of HDL subfractions.
Remodeling of HDL
Remodeling of HDLs involves the interaction with multiple proteins and enzymes over the 4–5 day life cycle. 39 The role of these key proteins and enzymes and their potential mechanisms during HDL subfractions remodeling will be discussed. Hydrolysis of lipids of HDL is mediated by various lipases [hepatic lipase (HL), endothelial lipase (EL) and lipoprotein lipase (LPL)] and exchange of lipids by the phospholipid transfer protein (PLTP) and CETP, finally forming distinct subfractions differing in size, composition, and functional properties.
Hepatic Lipase (HL) and Endothelial Lipase (EL)
The hydrolysis of TG and PL by the plasma lipases HL and EL is crucial for HDL remodeling. By hydrolyzing TG and PL, HL and EL convert lipid-rich large HDL2 to smaller, protein-rich HDL3, thereby destabilizing HDLs and leading to the shedding of lipid-free ApoA-I or lipid-poor preβ-HDL from the particle surface, which is preferentially cleared by the kidneys (Figure 2A).
40
Analysis of HL transgenic rabbits suggested that HL reduces the size of α-migrating HDL and increases the rate of catabolism of ApoA-I.
41
In contrast, ApoE-/- mice deficient in HL or EL exhibited elevated levels of large HDLs and reduced AS. Hence, HL and EL are thought to be negative regulators of plasma HDL-C levels, with a positive correlation with small HDL and a negative correlation with large HDL.
42
However, Silbernagel et al. found that reduced HL activity may be associated with high LDL-TG and lead to increased CVD risk in an epidemiological and Mendelian randomized study.
43
More work is required regarding the impact of HL and EL on HDL remodeling and ASCVD. Schematic representation of HDL subfractions remodeling by the action of HL/EL, LPL, PLTP, and CETP. (A) Hepatic lipase (HL) and endothelial lipase (EL) hydrolyze phospholipids (PL) and triglyceride (TG) of large HDL2, forming smaller HDL3 and lipid-poor apolipoprotein A-I (ApoA-I). (B) Lipoprotein lipase (LPL) hydrolyzes TG in triglycerides-rich lipoproteins (TGRLs), releasing superficial PL and free cholesterol (FC) to small, dense, lipid-poor HDL3, and discoidal particles. (C) Phospholipid transfer protein (PLTP) enhances the net transfer of PL from very-low-density lipoproteins (VLDL) and low-density lipoproteins (LDL) to HDL and converts HDL3 into HDL2 and preβ-HDL. (D) Cholesteryl ester transfer protein (CETP) facilitates the transfer of cholesteryl esters (CEs) from spherical HDL into TGRLs to exchange for TG. The remodeling of HDL by different proteins and enzymes is shown as broken lines.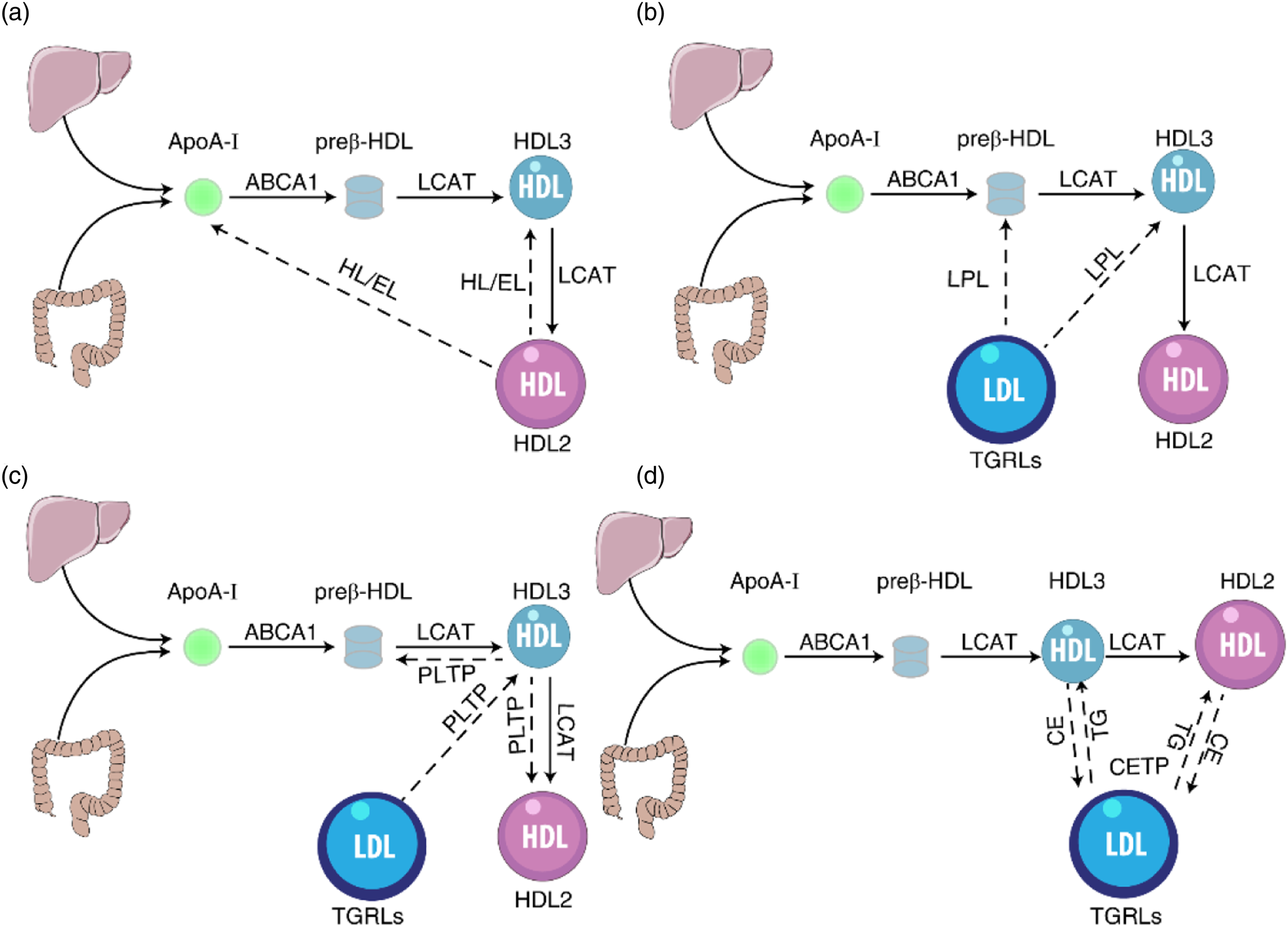
Lipoprotein Lipase (LPL)
HDL can also be remodeled through material transport with other lipoproteins. LPL, a major enzyme on the vascular endothelial surface, can decompose endogenous TG in circulating triglycerides-rich lipoproteins (TGRLs) such as low-density lipoproteins (LDL), very-low-density lipoproteins (VLDL), and chylomicrons (CM). Catalyzed by LPL, VLDL is transformed to LDL, and CM is utilized to produce de novo HDL. In the process of hydrolyzing TGRLs, their TG are lipolyzed to free fatty acids by LPL at the surface of the endothelium, leading to the shrinkage of hydrophobic lipoprotein core and the production of smaller-sized, higher-density remnant TGRLs. Excess molecules of the surface monolayer are shed from the particles and transferred to HDLs, such molecules include surface apolipoproteins, PL, and FC. Small, dense, lipid-poor HDL3, and discoidal particles represent preferential acceptors for released molecules during TGRLs lipolysis (Figure 2B), which enhances LCAT-mediated formation of HDL-cholesteryl esters (HDL-CEs), facilitating the conversion of HDL3 into HDL2. 44 As a result, there is a precursor-product relationship between TGRLs surface components and HDL2 that goes through the intermediate HDL3. 45 A high level of HDL2 reflects the metabolic efficiency of LPL to TGRLs, whereas a low level of HDL2 indicates inadequate LPL activity. 46 Kang et al. found that the supplementation of nordihydroguaiaretic acid (NDGA, an LPL inhibitor) increased the proportion of small HDL (HDL3a+3b + 3c) and decreased the proportion of large HDL (HDL2a+2b). 47 These findings suggest that LPL regulates HDL size and HDL subfractions.
Phospholipid Transfer Protein (PLTP)
PLTP, a member of the lipid transporter family, is mainly expressed in the liver and small intestine, which performs two major functions in HDL remodeling: promoting the net transfer of PL from TGRLs to HDL and increasing the formation of mature HDL. PLTP is considered to be an HDL conversion factor, 48 which generates large fused HDL2 particles and preβ-HDL by promoting the remodeling of HDL3 (Figure 2C). Therefore, PLTP can participate in the release of lipid-poor ApoA-I via the fusion of HDLs. Lipid-poor ApoA-I acts as an effective cholesterol acceptor and involves in cellular cholesterol clearance. In the PLTP-deficient mice, HDL-C and ApoA-I markedly decreased but reduced AS, this may be attributed to reduced hepatic VLDL secretion and blocked lipid transfer from VLDL to HDL. 49 It has also been reported that deficiency of PLTP leads to alleviation of AS in LDLR-/- mice. Compared with wild-type mice, transgenic mice overexpressing PLTP found that the preβ-HDL is significantly increased. 50 Interestingly, PLTP overexpression also lead to a decrease in HDL-C, but the reason remains unclear. Furthermore, PLTP not only affects the size and level of HDLs but also changes its inflammatory index. 51 Gautier found that PLTP plays a key role in regulating immune responses and is directly related to a wide range of inflammatory diseases, including bacterial infections and sepsis. 52 In general, PLTP is a proven AS risk factor in animal models. Inhibition of PLTP may be a treatment for dyslipidemia and CVD, but whether there are more advantages than disadvantages in patients with ASCVD is worthy of further study.
Cholesteryl Ester Transfer Protein (CETP)
CETP is a hydrophobic glycosylated protein principally involved in HDL remodeling. CETP mediates the transfer of CE from spherical HDL into TGRLs to exchange for TG, 6 leading to the formation of TG-enriched HDLs and transformation of the large HDL2 into small HDL3 (Figure 2D). TG-enriched HDLs are then easily hydrolyzed by HL. The concerted action of CETP and HL promotes a reduction in HDL size. Thus, CETP levels are associated with increased small preβ1-HDL and decreased large HDL subfractions (HDL2a and HDL2b), suggesting that CETP may be a limiting factor for RCT and HDL maturation. The mice overexpressing CETP showed a significant decrease in HDL-C and ApoA-I, and an increase in preβ-HDL levels that are susceptible to AS. 53 Inhibition of CETP activity in humans is associated with increased plasma levels of HDL-C and decreased levels of small preβ1-HDL particles. 54 The effect of CETP on AS is relatively complex, but it is widely believed to be a positive correlation with the incidence of CV risk. 55 Consequently, CETP inhibitors are constantly being developed and utilized, which can increase HDL-C levels and change the size of HDLs. But some CETP inhibitors failed to increase atheroprotection in clinical studies. Perhaps CETP inhibitors exert certain anti-atherogenic effects in high levels of low-density lipoprotein cholesterol (LDL-C) conditions. There is, therefore, a great need for further studies to precisely define the role of the CETP in ASCVD.
Metabolism of HDL
HDL is metabolized by many proteins after remodeling in plasma. The HDL-C can be removed through different pathways by the action of specific cell receptors [scavenger receptor class B type I (SR-BI) and LDL receptor (LDLR)]. It was estimated that 66% of the CEs of HDL are transferred to LDL/VLDL via CETP, and subsequently, LDL/VLDL containing HDL-derived CEs is actively transported into hepatocytes by endocytosis to form the LDL/LDLR complex. 56 The complex enters the cytoplasm through the invagination of the cell membrane to form vesicles, and the lipid component of LDL/VLDL is hydrolyzed by enzymes in the lysosome thereby indirectly removing HDL-CEs. 57 Approximately 33% of CEs in HDL are delivered to the liver for selective uptake by SR-BI. 56 Hepatic cholesterol can be excreted into the bile, which is either reabsorbed by the intestine or ultimately excreted in feces. Besides, plasma lipoprotein-derived cholesterol can be secreted into the intestinal lumen, an alternative nonbiliary process known as transintestinal cholesterol excretion (TICE). 1 Regardless of the pathway through which it is metabolized, the lipid-poor HDL eventually is released back into the circulation, where it may undergo another round of direct RCT, or may undergo metabolism and clearance by the kidneys. Recent evidence has suggested that HDL can be integrated into the lipid bilayer of cells, which perhaps is a novel method for HDL clearance. 58
HDL Subfractions and Atherosclerotic Cardiovascular Diseases
Biological activities of HDL subfractions. a
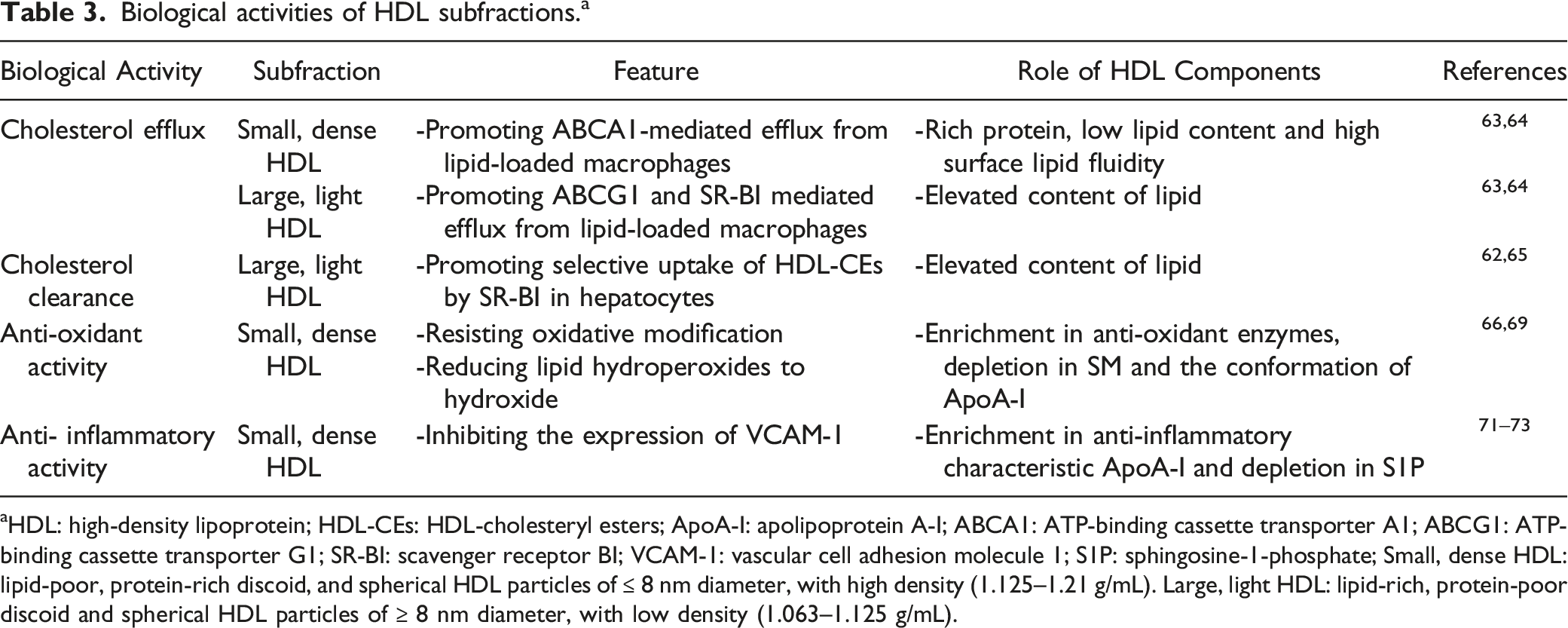
aHDL: high-density lipoprotein; HDL-CEs: HDL-cholesteryl esters; ApoA-I: apolipoprotein A-I; ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; SR-BI: scavenger receptor BI; VCAM-1: vascular cell adhesion molecule 1; S1P: sphingosine-1-phosphate; Small, dense HDL: lipid-poor, protein-rich discoid, and spherical HDL particles of ≤ 8 nm diameter, with high density (1.125–1.21 g/mL). Large, light HDL: lipid-rich, protein-poor discoid and spherical HDL particles of ≥ 8 nm diameter, with low density (1.063–1.125 g/mL).
HDL Subfractions and Reverse Cholesterol Transport
The RCT pathway mainly consists of three processes: 1. Cholesterol efflux from peripheral cells to plasma HDL; 2. LCAT-mediated esterification of cholesterol and remodeling of HDLs; 3. Direct and indirect clearance of HDL-CEs. 62 Briefly, the RCT process dominated by HDL represents the maturation and metabolic process of HDL. The cholesterol efflux capacity (CEC) is a key factor in the initiation step of the RCT, and is related to HDL subfractions and particle size. Du pointed out that higher density HDLs, such as HDL3, are the most efficient acceptors of cholesterol efflux, and ABCA1 mainly mediates cholesterol efflux to HDL3b, HDL3c, and lipid-free ApoA-I. 63 These may account for the protein-rich and negatively charged phospholipid of HDL3. 64 Pownall and Ehnholm proposed that these antiatherogenic effects of HDL are attributed to phosphatidylcholine, because it is the only agent that is shown to promote the regression of experimental AS. 65 HDL3 with rich phosphatidylcholine act as cholesterol shuttles and initiate cholesterol removal from membranes and transport it to the larger HDLs, which serve as passive transporters. 64 Lipid-rich HDL2 is a more efficient ligand for cellular uptake of HDL-CEs mediated by SR-BI than small, lipid-poor HDL in hepatocytes. 66 Some evidence also suggests that an inverse association between large α1 HDL levels and AS.67,68 However, this association may not necessarily be interpreted as a direct consequence of the protective properties of large HDL subfractions, as it may reflect the cardioprotection provided by metabolic processes leading to the formation of large α1 HDL. Overall, these results indicate that small, dense HDLs are more potent in promoting cholesterol efflux based on phospholipid content, whereas large HDLs are more effective based on particle number.
HDL Subfractions and Anti-oxidant Activity
The anti-oxidant properties of HDL in vivo can be divided into direct and indirect effects. HDL can protect lipid and protein moieties of LDL from oxidative damage caused by free radicals. It has been reported that small, dense HDL3 may be superior to large, light HDL2 in terms of their capacity to resist oxidative modification, 69 which can be attributed to the enrichment of some anti-oxidant enzymes in HDL3, such as paraoxonase 1 (PON1), platelet-activating factor-acetyl hydrolase (PAF-AH) that hydrolyze short-chain oxidized phospholipids. Another HDL enzyme possessing anti-oxidant activity is LCAT, and the sphingomyelin/phosphatidylcholine (SM/PC) ratio of HDL might influence enzymatic activities. 70 Furthermore, HDL3 is more effective in reducing lipid hydroperoxides to hydroxide by inactivating lipid hydroperoxides, the major product of LDL lipid peroxidation, compared with HDL2. It seems that the major protein component of small, dense HDL can be attributed to ApoA-I which might facilitate the redox reaction between the methionine residues and the lipid hydroperoxides in the structure. Other unique proteomes of HDL3 may influence its anti-oxidant activity, which is highly correlated with the presence of ApoA-Ⅱ, ApoA-Ⅳ, ApoE, ApoJ, and ApoM. 14 Inhibition of oxidative stress may be achieved indirectly via other functions of HDL, such as induction of cholesterol efflux and anti-inflammatory functions. In summary, the anti-oxidant activity of HDL subfractions is related to the presence of several apolipoproteins and enzymes with anti-oxidant properties and the lipid components can significantly modulate anti-oxidant activities displayed by the protein components.
HDL Subfractions and Anti-inflammatory Activity
HDL exerts anti-inflammatory activities through multiple mechanisms, including inhibition of adhesion molecule expression in endothelial cells, reduction of monocyte adhesion to the endothelium, inhibition of monocyte and neutrophil activation, and prevention of neutrophil infiltration into the arterial wall. 14 Ashby et al. demonstrated that HDL3 was markedly superior to HDL2 in inhibiting the expression of vascular cell adhesion molecule 1 (VCAM-1) in human umbilical vein endothelial cells (HUVECs) incubated with tumor necrosis factor-α (TNF-α). 71 Perhaps the anti-inflammatory characteristic ApoA-I enriched in small HDL basically explains this phenomenon. Besides, HDL3 can inhibit TNF-α induced sphingosine-1-phosphate (S1P) production in endothelial cells, which enhances sphingosine kinase activity and adhesion protein expression. 72 Scientists also found spherical HDLs have more anti-inflammatory activity than discoid HDL. 73 Perhaps the phospholipid content of HDL subfractions may contribute to these observations, which may interfere with the ability of HDL subfractions to inhibit the expression of adhesion molecules. However, the composition and structure of HDL are altered in the setting of systemic inflammation, it may lose its protective effects and even acquire deleterious functions, which is called dysfunctional HDL. These dysfunctional HDLs can cause impaired cholesterol efflux capacity, diminished antioxidant effects, and increased pro-inflammatory effects. Therefore, understanding the characteristics of dysfunctional HDL may provide ideas for exploring new diagnostic and therapeutic approaches for ASCVD.
Pharmacological Intervention to Modulate HDL Subfractions
A wide range of retrospective and prospective epidemiological studies have consistently demonstrated that HDL is inversely correlated with the incidence of atherosclerotic disease, and HDL is incorporated in clinical guidelines as one of the primary parameters for assessing CV risk. 74 We reviewed the current drugs targeted to regulate HDL, which usually affect the concentration of specific HDL subfractions.
Niacin
Effects of classical lipid-regulating drugs on HDL-C and HDL subfractions. a
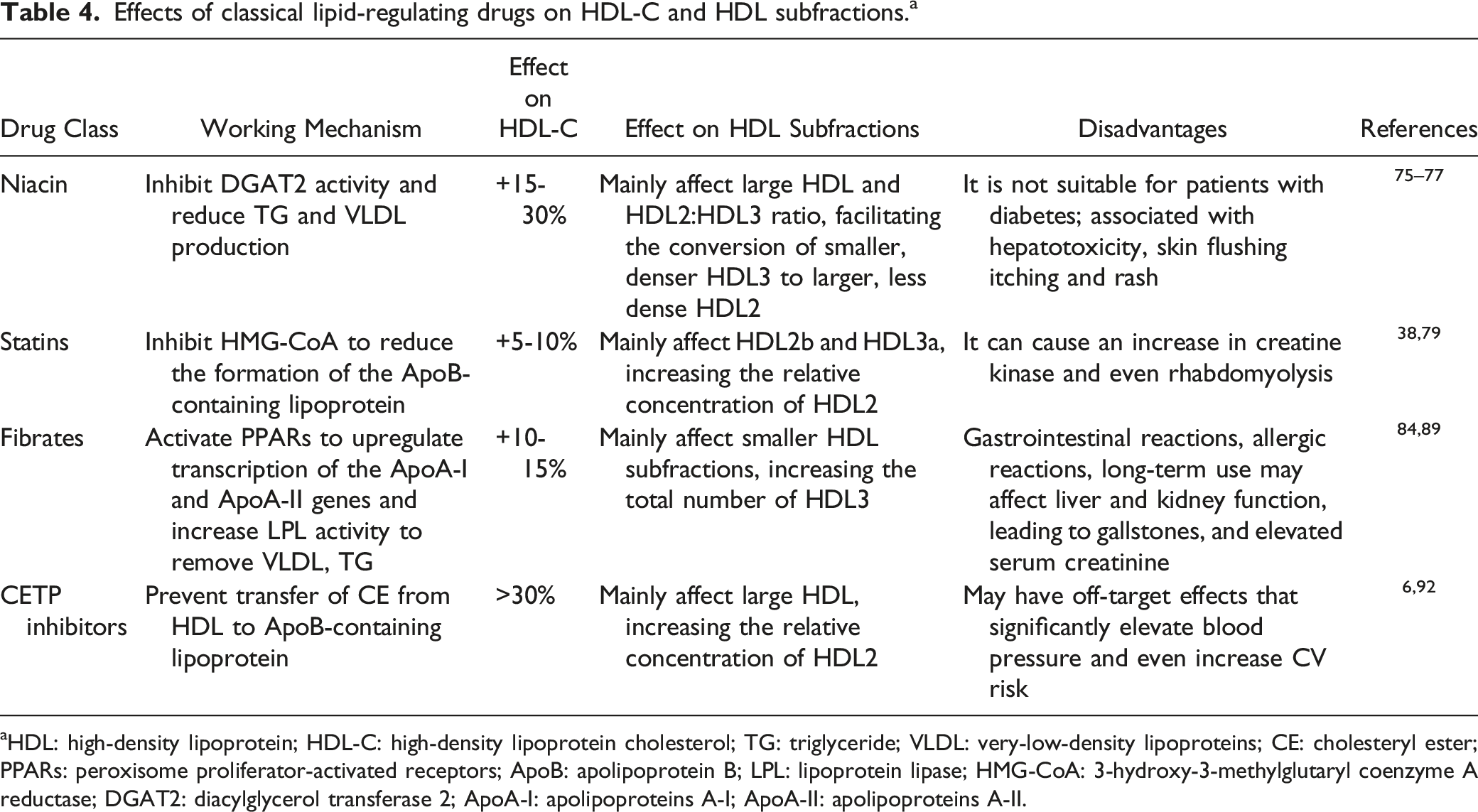
aHDL: high-density lipoprotein; HDL-C: high-density lipoprotein cholesterol; TG: triglyceride; VLDL: very-low-density lipoproteins; CE: cholesteryl ester; PPARs: peroxisome proliferator-activated receptors; ApoB: apolipoprotein B; LPL: lipoprotein lipase; HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A reductase; DGAT2: diacylglycerol transferase 2; ApoA-I: apolipoproteins A-I; ApoA-Ⅱ: apolipoproteins A-Ⅱ.
Statins
Statins disrupt the biosynthesis of cholesterol by inhibiting the3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA). Statins are the most widely used lipid-lowering drugs owing to an active reduction in LDL-C levels. While the primary benefit of statins relates to their ability to lower LDL-C levels, a secondary effect shows a small increase in HDL-C levels. Statins typically increase HDL-C by 5-10%. 38 Statins affect the HDL profile through two complementary mechanisms: inhibition of CETP and reduction in the production of ApoB-containing lipoprotein particles. 79 However, statin treatment does not completely normalize the risk of CVD associated with dyslipidemia and the effect of statins on HDL-C varies by dose and formulation. 80 The elevated HDL-C responses observed from certain statins may not parallel their dose-dependent LDL-C lowering responses. 81 For example, the response of atorvastatin to elevated HDL-C appeared to decline with increasing dose, by contrast, rosuvastatin showed a more consistent dose-response relationship in terms of both LDL-C reduction and HDL-C elevation. 81 Of note, the slight increase in HDL-C in statin-treated patients appears to be mainly associated with an increase in HDL2 rather than HDL3. 82 This may be one reason why statin therapy does not seem to be effective in reducing the risk of ASCVD in patients with low HDL-C levels. 83
Fibrates
Fibrates activate the intrahepatic nuclear peroxisome proliferator-activated receptors (PPARs) to modulate protein transcription of the ApoA-I and ApoA-II genes. 84 Activation of PPARs ultimately increases plasma LPL activity, resulting in the observed changes in lipid fractions. Conversely to the aforementioned drugs, fibrate treatment may promote increases in HDL3. The Veterans Affairs High-Density Lipoprotein Intervention Trial (VA-HIT) study showed that gemfibrozil decreased coronary artery disease mortality, which may be attributed to the significant reduction of the HDL3-C ratio.85,86 In a study of patients with CVD, fenofibrate increased plasma ApoA-II levels by 32%, consistent with a greater effect on smaller HDL. 87 Although fibrate improves lipid profiles, unfortunately, these increases in HDL3 are relatively low and may be insufficient to reduce the risk of CV events. 88 Nevertheless, there is no denying the effectiveness of fibrate monotherapy in the treatment of hypertriglyceridemia. In addition to fibrate monotherapy, combination of a fibrate with a statin is an option for the patients with combined dyslipidemia and diabetes mellitus who present with atherogenic dyslipidemia (low HDL-C and elevated TG levels).89,90 However, this combination therapy is rarely used in clinical practice because of the liver and muscle toxicity as well as impact on renal function. 91 The pathophysiological basis and clinical implications of fibrate-related effects need further investigation.
Cholesteryl Ester Transfer Protein (CETP) Inhibitors
Some of the most promising drugs for raising HDL are CETP inhibitors, which promote cholesterol efflux and raise the level of large HDLs, is the common mechanism for the action of niacin, statins, and fibrates on HDL-C effects. 82 Clinical trials of four CETP inhibitors have been completed: torcetrapib, dalcetrapib, anacetrapib, and evacetrapib, mostly with disappointing results.92–94 Indeed, the meta-analysis concluded that current CETP inhibitors did not reduce ASCVD mortality. 95 However, the Randomized Evaluation of the Effects of Anacetrapib through Lipid Modification (REVEAL) trial seems to provide promising results. The REVEAL trial is a randomized trial investigating the effects of adding anacetrapib to an effective LDL-lowering treatment with atorvastatin, in which the inhibitor anacetrapib significantly raised HDL-C and reduced LDL-C, with a protective effect on major coronary events. 96 The decrease in the concentration of LDL-C can be explained not only by the block of the transfer of CEs from HDL to LDL. It may also be that anacetrapib reduces plasma levels of the proprotein convertase subtilisin kexin type 9 (PCSK9), which is one of the major regulators of LDLR. 97 Although the REVEAL trial has shown that anacetrapib could reduce the risk of major coronary events, as it is highly lipophilic and accumulated in adipose tissue during continued dosing 98 ; further regulatory approval is not be sought.
Despite the development of effective HDL-raising drugs, large-scale clinical trials showed disappointing results with no significant reduction of clinical CV events. This implies that much of the current pharmaceutical-based therapies have causal effects on HDL-C, but are not HDL function-targeted. This also suggests that focusing on improving HDL quality rather than just increasing HDL quantity may be the next target for future therapies. Many novel therapies primarily targeting HDL subfractions, that are reconstituted HDL (rHDL), delipidated HDL, ApoA-I mimetics, ApoA-I up-regulators, LCAT activators, and SR-BI agonists, also play unique roles in ASCVD.99,100 Notably, rHDL is a complex of ApoA-I with a variety of phospholipids and to a large extent functions similarly to natural HDL. Some studies have indicated that rHDL could exert a protective effect by improving RCT or by absorbing beneficial proteins. 101 Therefore, we propose that rHDL therapy may be one of the most promising new approaches to targeting specific HDL subfractions, thereby improving function and reducing CV risk.
Conclusion
Raising HDL-C is generally thought of as a promising strategy to reduce the risk of ASCVD, but emerging evidence suggests that the atheroprotective role of HDL is not simply estimated by the plasma HDL-C levels. Previous approaches aimed at improving ASCVD risk have focused on pharmacological increases in HDL-C concentrations. These approaches have largely failed, partly because of the inherent complexity of HDLs. Given that the function of HDL is attributed to the heterogeneity of HDL subfractions, it becomes easily understandable that the remodeling of HDL subfractions may be a critical process for the treatment of ASCVD. We recommend that future research focuses to a greater extent on the remodeling of HDL subfractions and the functionality of HDL subfractions.
Footnotes
Acknowledgments
We thank all collaborators of this work and the grant support of Natural Science Foundation of Hunan Province and Scientific Research Project of Health Commission of Hunan Province.
Author Contribution
Yaling Zhang and Shiyu Luo equally contributed to this manuscript.
YZ and SL designed and drafted the manuscript. The rest of the authors participated in analysis and interpretation of data and revising it critically for important intellectual content. All authors approved the final version of the manuscript and agreement to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Hunan Province (No 2016JJ3109, 2021JJ30599, and 2021JJ30623) and Scientific Research Project of Health Commission of Hunan Province (No D202302068763 and D202302067797).