
Abstract
Immune checkpoint inhibitors (ICIs) are specific monoclonal antibodies directed against inhibitory targets of the immune system, mainly represented by programmed death-1 (PD1) ligand-1 (PD-L1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4), thus enabling an amplified T-cell–mediated immune response against cancer cells. These drugs have significantly improved prognosis in patients with advanced metastatic cancer (e.g., melanoma, non-small cell lung cancer, renal cell carcinoma). However, uncontrolled activation of anti-tumor T-cells could trigger an excessive immune response, possibly responsible for multi-organ damage, including, among others, lymphocytic myocarditis. The incidence of ICIs-induced myocarditis is underestimated and the patients affected are poorly characterized. The diagnosis and management of this condition are mainly based on expert opinion and case reports. EKG and ultrasound are tests that can help identify patients at risk of myocarditis during treatment by red flags, such as QRS complex enlargement and narrowing of global longitudinal strain (GLS). Therapy of ICI-related myocarditis is based on immunosuppressors, monoclonal antibodies and fusion proteins. A future strategy could involve the use of microRNAs. This review considers the current state of the art of immune-related adverse cardiovascular events, focusing on histological and clinical features, diagnosis and management, including current treatments and future pharmacological targets.
Introduction
Immune-checkpoint inhibitors (ICIs) have significantly improved clinical outcomes in cancer treatment and are progressively being used both in early and metastatic disease as single or combined anticancer therapy. 1 Nevertheless, due to the increased immune response, ICIs could induce immune-mediated adverse events with systemic involvement, sometimes responsible for severe or even fatal complications.
Immune-related cardiovascular (CV) damage has been reported in different case series. However, the lack of a defined diagnostic work-up and specific symptoms could be responsible for a high rate of misdiagnosis. 1 Cardiac involvement can be inflammatory, manifesting as pericarditis, myocarditis, vasculitis, or noninflammatory, including left ventricular (LV) dysfunction, takotsubo syndrome, myocardial infarction, and arrhythmias.2,3 Cardiovascular immune-related adverse events (CV-irAEs) appear more frequently in patients treated with cytotoxic T-lymphocyte antigen-4 (CTLA-4) blockers as compared with programmed death-1 (PD-1) inhibitors, 4 and ICIs-combination seems to have higher incidence and death rate as compared with ICI monotherapy.5–7
The largest clinical characterization of CV-irAEs is the pharmacovigilance analysis of the VigiBase World Health Organization (WHO) database. 6 In more than half of patients (53%), cardiac toxicity occurred within 4 weeks after starting ICIs-combination, whereas it occurred in 17% after the first dose and in 34% of patients at 4-months with ICI monotherapy.6,8 Myocarditis was the most common adverse event. It generally occurs after the first infusion, with poor outcomes in 50% of cases and a reported median time of symptom onset between 34 and 65 days.6,8 However, the occurrence of this adverse event could be delayed. For example, Yamaguchi et al. described a case of fulminant myocarditis occurring 1 year after the start of immunotherapy. 9 In addition, myocarditis was more frequent and severe in patients treated with the combination of anti-PD-1 and anti-CTLA-4 than in those treated with anti-PD-1 in monotherapy. 10
Pathophysiological mechanisms
The most accredited hypotheses of the pathophysiology of CV-irAEs are the “shared antigen” and “epitope mimicry” theories; muscle-specific antigens, such as desmin, troponin, titin, and myosin heavy chain are often detected in cancer cells. 11 As most self-antigens are intracellular proteins, surface exposure is likely to occur after cardiac damage induced by a previous inflammatory process.11,12 Thus, hyperproliferative T-lymphocytes and macrophages induced by ICIs therapy could infiltrate the cardiac muscle, potentially causing myocardial damage.11–15 Cardiac-myosin-specific autoimmune T cells may be present in mouse hearts expressing PD-1 in naïve conditions, thus triggering PD-1 inhibitor-induced myocarditis.
The risk of autoimmune T-cell-mediated myocarditis through heart antigen-specific effector T cells may be increased by genetic and hereditary deficiencies of checkpoint molecules, such as PD-1, PD-L1, lymphocyte-activation gene-3 (LAG-3), and CTLA-4.
16
Especially, PD-L1 plays a key role in controlling immune-mediated cardiac injury, considering that its up-regulation on cardiac endothelial cells (EC) was preclinically observed in response to acute lymphocytic myocarditis.
16
The first published description of the PDL-1 molecule found high levels in heart tissue in mouse model.
17
Subsequent studies confirmed that PD-L1 expression could be activated on cardiac EC by interferon-gamma (IFN-γ), a pro-inflammatory cytokine, in an autocrine and paracrine manner, in order to protect heart tissue by T-cell inflammation damage.
18
This concept has been further explained with an in vivo model of myocarditis, determined by adoptively transferred T-cell receptor (TCR) transgenic heart antigen cytotoxic-T lymphocyte (CTL), showing that PD-L1 expression is clearly up-regulated on cardiac cells by CTL activation in the heart.18,19 Furthermore, although IFN-γ is responsible for the up-regulation of class I major histocompatibility complex (MHC-I) expression on EC surface, the induced PDL-1 expression decreased EC ability to activate cytotoxic T-lymphocytes through class I MHC-restricted antigen presentation, and acquired an increased resistance to antigen-specific CTL killing (Figure 1).
18
Molecular mechanisms of ICI-related myocardial injury. CD4 = cluster of differentiation 4; CD8 = cluster of differentiation 8; CTLA-4 = cytotoxic T-lymphocyte antigen-4; LAG 3 = lymphocyte-activation gene-3; PD-1 = programmed death-1; PD-L1 = programmed death-1 ligand.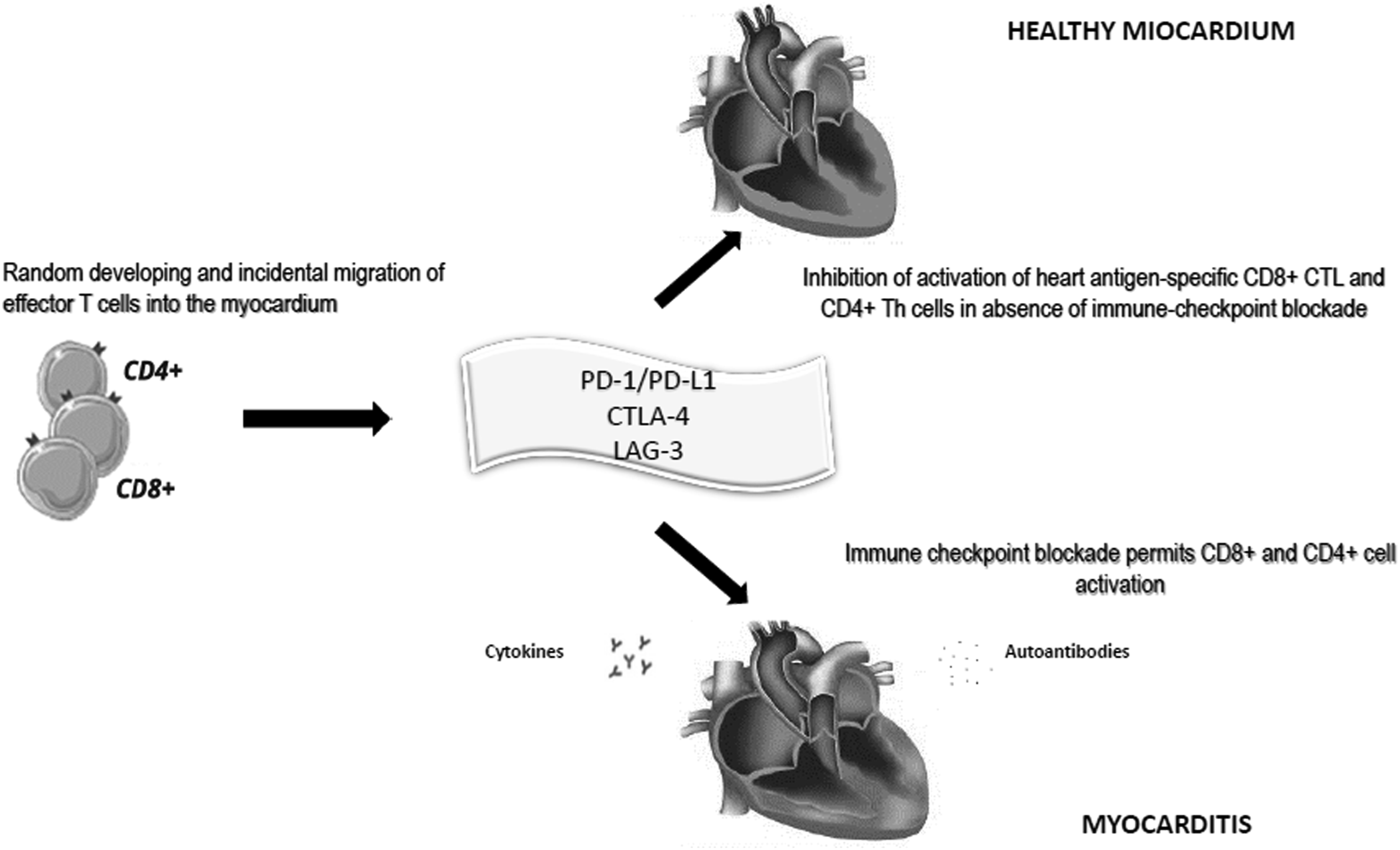
Preclinical evidence of ICI-induced cardiotoxicity derives from mouse models deficient in PD1 and PDL-1, even though with strain-specificity.20,21 PD-L1−/− Murphy Roths large (MRL)- spontaneous lymphoproliferative mutation (Faslpr) mice develop a fulminating, fatal autoimmune myocarditis that is similar to human autoimmune myocarditis while PD1-deficient laboratory-bred, albino strain (BALB/C) mice exhibit dilated cardiomyopathy with pathological cardiac remodeling of the myocardium and severely reduced LV function, associating with autoantibodies specific for cardiac troponin I (cTnI). The autoantibodies bind to TnI on the surface of cardiomyocytes and stimulate voltage-dependent l-type Ca++ currents, leading to cardiomyocyte necrosis and arrhythmias. 20 This disease phenotype could be also reproduced by injecting wild-type BALB/c mice with monoclonal anti-TnI antibody, 12 but with no evidence of myocardial inflammation or T-cell-mediated cardiac injury.
Histological analyses of patients and monkey models have suggested that T-helper (Th)1/Th2 cell imbalance and T follicular helper (Tfh) cells play an essential role in developing myocarditis, leading to a predominant CD4+/CD8+ T-lymphocytes infiltration rather than CD68+ macrophages.22–26 Xue et al. observed that CD4+ chemokine receptor (CXCR)-5+ Tfh cells and CD19+ B cells were both significantly higher in spleen and myocardial tissue in an experimental model of autoimmune myocarditis. 27 The researchers reported high serum levels of Interleukin (IL)-21, chemokine ligand (CXCL)-13, and myosin antibodies, 27 supporting previous evidence about the expression of multiple chemokine receptors and ligands (CXCR3-CXCL9/CXCL10 and CCR5/CCL5) that upregulate T-cell production of tumor necrosis factor (TNF)-alpha, granzyme B, and IFN-y, inducing cell death and cardiac injury.28–30
B7 may enhance memory T cell activation- Cluster of Differentiation (CD) 28 co-stimulation; therefore, blocking CTLA-4 could enhance reactivation of previously generated heart-antigen specific memory T cells. 31 This could, in part, explain the higher toxicity of ICI-combination.
Direct T-cell-mediated cytotoxicity causes macrophage infiltration and fibrosis of the working myocardium and conduction system, causing bradyarrhythmias and tachyarrhythmias.32,33 Inflammatory-induced oxidative stress leads to cardiomyocyte necrosis, with subsequent electrical and structural remodeling; chronic inflammation links to autonomic dysfunction, with sympathetic overactivation and a decline in parasympathetic function; autoantibody-mediated inhibitory effects exacerbate cardiac arrhythmias. 34 Finally, cytotoxic T-cell response could spread to the pericardium by contiguity, even though pericardial toxicity is often reported after radiotherapy that facilitates cross-reactivity mechanisms with tumor shared antigens. 35
Human reports of histological features of ICI-induced myocarditis are lacking, limited to single case reports or small series. This evidence showed that myocardial infiltrate is predominantly lymphocytic or lymphohistiocytic, notwithstanding few of them have reported the presence of eosinophils and, in one case, also giant cells. 36 Globally, there are pathologically different forms of myocarditis with distinct clinical evolutions, often with marked similarities to cardiac allograft rejection. 36 Champion et al. analyzed histological correlations between ICI-induced myocarditis and cardiac allograft rejection and evidenced that the heterogeneous composition of the inflammatory cell infiltrate is associated with different outcomes. They classified myocarditis as high grade (H-G) or low grade (L-G) based on the number of T-cell activated CD3+ lymphocytes greater or less than 50/high-power field. Higher densities of CD3+ and CD8+ cells are typical of H-G myocarditis and cardiac allograft rejection; CD68+ macrophages are more evident in H-G myocarditis compared with L-G myocarditis and rejection; higher CD68+/CD3+ ratio and density of PD-L1 on macrophages and myocytes are typical of both grades, unlike rejection. Finally, H-G is characterized by significantly more myocyte necrosis and thus could be considered necrotizing myocarditis analogous to acute eosinophilic and giant cell myocarditis, even if with less eosinophilic infiltration. 36 These histopathologic features may, in part, explain the different spectrum of clinical manifestations and outcomes, the current choice of treatment and the limitations in terms of efficacy.
Diagnosis
The diagnosis of ICI-related myocarditis is often a clinical challenge. A comprehensive clinical evaluation is required to obtain a definitive diagnosis. Clinical history and physical examination should be carefully performed to rule out other cardiac causes for the patient’s clinical presentation, such as viral and autoimmune cardiac disease, infectious myocarditis, or myocardial infarction. 6
ICI-related myocarditis could manifest with a wide spectrum of symptoms, such as myasthenia gravis-like symptoms, heart failure, myositis, cardiac arrhythmias, pericardial disorders, and pneumonitis.37,38 The clinical presentation often mimics that of patients with conduction abnormalities or acute coronary syndrome. These conditions should be excluded before starting the diagnostic workup for ICI-induced myocarditis.39,40
In the setting of ICI-associated myocarditis, ECG frequently shows conduction abnormalities (89%), atrial fibrillation (30%), ventricular tachyarrhythmia (27%), and atrioventricular conduction disorders (17%). However, ECG lacks sensitivity and specificity for the diagnosis: in a recent multicenter study Zlotoff et al. exclusively identified an association between QRS duration >120 msec, ICI myocarditis, and increased major adverse cardiac event (MACE) risk. 41
Despite limited evidence, the myocardial troponin (Tn) assay as an index of cell damage has been proposed as a biomarker to monitor cardiac involvement, both at baseline (i.e., before drug infusion) and during treatment. 42 However, the increase in Tn levels could be triggered by numerous factors like pre-existing subclinical CV disease, prior or concomitant cardiotoxic therapy, or supply-demand mismatch, leading to inappropriate interruption or delay of potentially life-saving therapy. 42 Petricciuolo et al. suggested that a baseline high-sensitivity Tn T (hs-TnT) >14 ng/L could be a good predictor for CV death, stroke, or transient ischemic attack, pulmonary embolism, and cardiac involvement progression at 3 months. 43 On the other hand, Lee Chuy et al. did not demonstrate a clear efficacy of serial measurements of hs-TnI before each infusion in predicting a potential ICI-induced acute cardiotoxicity. 44
Moreover, it is possible to observe trends toward higher serum troponin levels and shorter interval from the first ICIs treatment in the high-grade myocarditis group compared with the low-grade group. Considering that all the patients with high-grade myocarditis have a worse prognosis compared with low-grade, troponin kinetics may have a significant prognostic value.
36
Specifically, in symptomatic patients, elevated cTnI levels represent an important marker for autoimmune myocarditis with high sensitivity and specificity.
39
At the same time, cTnT and creatine kinase (CK) could also be elevated in myositis or myasthenia gravis42,44; especially ICI-induced myositis may represent an alternative non-cardiac cause of cTnT elevation, in absence of any cardiac symptoms, clinical and echocardiographic signs of congestion or rhythm abnormalities, as recently pointed out by Ruperti-Repilado et al.
45
Mahmood et al. observed that patients who experienced a MACE had high baseline, peak, and discharge cTnT values. In addition, a discharge level of cTnT ≥1.5 ng/mL was associated with an increase in the likelihood of a MACE.
39
On the other hand, brain natriuretic peptide (BNP) and NTerminal-proBNP (NT-proBNP) are thought to be less specific, but their use could be considered for monitoring disease course (Figure 2).
46
Diagnostic pathway of patients treated with immune checkpoint inhibitors and suspected myocardial injury. ACS = acute coronary syndrome; BNP = brain natriuretic peptide; CK-MB = creatin phosphokinase MB; CPK =creatin phosphokinase; cTnI = troponin I; cTnT = troponin C; ECG= electrocardiogram; EMB = endomyocardial biopsy; LGE = late gadolinium enhancement; MRI = magnetic resonance imaging; pro-BNP-NT = N-terminal fragment of pro-BNP; T 2-STIR = short time inversion recovery.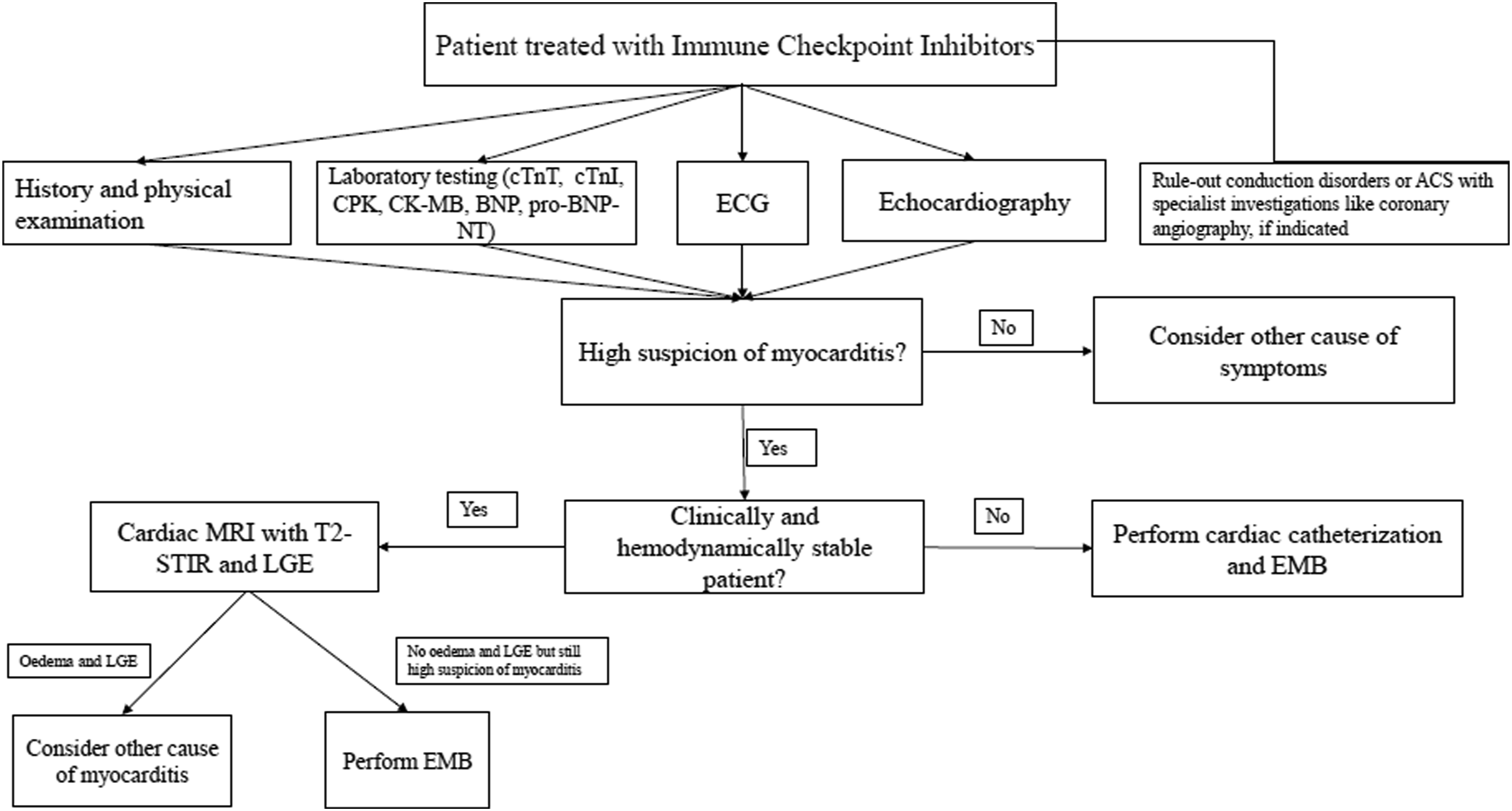
Myocarditis could lead to a MACE, such as cardiac arrest, cardiogenic shock and complete heart block, 47 but the diagnostic algorithm and risk stratification for cardiological sequelae are still debated. 48 Endomyocardial biopsy (EMB) is the gold standard for definitive diagnosis of myocarditis; 49 however, the procedure carries some risks and limitations. For example, inflammatory infiltration can be transient or focal; thus, biopsy sampling error could result in a false negative diagnosis. 49
In the context of myocardial injury, study by cardiac magnetic resonance (CMR) is currently recommended to identify myocardial areas with increased signal intensity in T2 sequences, edema with non-ischemic pattern, and fibrosis based on late gadolinium enhancement.50,51 If there are no abnormalities in the acute setting but clinical suspicion is strong, CMR can be repeated at least 2 weeks later.
The CMR, in addition to providing information on the global and regional contractility of the ventricles, allows for tissue analysis.47,50,51 Zhang et al. analyzed the association between CMR features and CV events among 103 patients with ICI-associated myocarditis, correlated with histopathological analysis of cardiac samples obtained by either EMB or post-mortem. 47 In this population, the fibrosis, analyzed by late gadolinium enhancement (LGE), and the edema, in the T2-weighted sequences, were poorly represented, in contrast to non-ICI-related myocarditis; moreover, gadolinium distribution patterns were highly variable (transmural, mesocardiac, subendocardial, etc.) and the probability of finding abnormalities at CMR was higher the later it was performed. 47 Therefore, the diagnosis is a multiparametric evaluation, in which a negative CMR should not rule out the diagnosis of myocarditis. 47 Similar conclusions have recently been observed by Cadour et al.; LGE was less frequent in patients with ICI-induced myocarditis than viral, mainly involving septal segments (16 of 33 patients [48%] vs 25 of 85 [29%]; P < .001) and midwall layer (11 of 33 patients [33%] vs 2 of 85 [2%]; P < .001), with septal LGE the only cardiac MRI predictor of MACE at 1 year (adjusted hazard ratio, 2.7 [95% CI: 1.1, 6.7]; P = .03). 51
In summary, in the case of clinically suspected myocarditis, cardiac catheterization with endomyocardial biopsy should be performed in unstable patients, while CMR is the first choice for clinically stable patients with the possibility to perform an endomyocardial biopsy in patients without LGE and with standard T2 sequences. However, coronary angiography is often required to exclude severe clinically significant obstructive lesions due to the overlapping presentation with an acute coronary syndrome.
Another aspect to consider is the correlation between myocardial damage and symptoms. In a prospective study, 22 patients who underwent CMR at baseline, before starting ICI, and at a 3-month follow-up, were analyzed, with new evidence of LGE in about 10% of patients, increased edema and reduced LV strain, in the absence of symptoms reported by patients. 52
Trans-thoracic echocardiography (TTE) could provide additional information on LV ejection fraction (LVEF), pericardial effusions and wall motion abnormalities, often evidenced in patients with myocarditis. Zhang et al. reported that more than half of the patients with ICI-induced myocarditis showed a preserved LVEF. 47 With the advancement of imaging methods, it is possible to analyze myocardial deformation by speckle tracking, an angle-independent analysis. 40 Global longitudinal strain (GLS) has proven to be an effective parameter for identifying early myocardial damage, prior to the reduction of LVEF. Several studies have demonstrated the efficacy of GLS in monitoring cardiotoxicity related to traditional cytotoxic chemotherapy, given its predictive positive value of subsequent decline in LVEF, but little is known about the effectiveness in monitoring ICI-related cardiotoxicity.53–55 Awadalla et al. retrospectively evaluated the use of GLS in patients who developed ICI-induced myocarditis. Compared with those without ICI-induced myocarditis, GLS at presentation was lower regardless of LVEF, the risk of subsequent MACE was higher with a lower GLS and directly proportional to the percentage reduction in GLS. Among the 26 patients with reduced LVEF who developed MACE, 23 had a GLS <13%, while among 25 patients with preserved ejection fraction and MACE, all of them had a GLS <16%. In conclusion, this study emphasizes that GLS is a valuable parameter for patient risk stratification. 40
Medical treatment
Clinical care necessitates an interdisciplinary approach, including oncologists, cardiologists, and coronary care unit. Patients receiving immunotherapies combination or other cardiotoxic antineoplastic drugs, such as tyrosine kinase inhibitors, or with a history of cardiac disease, have a higher risk of cardiotoxicity.56,57 Treatment strategies have three main objectives: to reduce the immune-related cardiotoxicity; to block inflammatory cascade with immunosuppressive drugs; to treat heart disease.
In case of mild myocardial damage, it is recommended to continue the ICI at least until the complication is resolved, with continuous clinical monitoring. In severe or life-threatening cases, ICI should be permanently discontinued (Figure 3). However, in case of latent or subclinical evolution with isolated troponin elevation, it could be reintroduced after careful cardiac monitoring and discussing with the patient the risk-benefit of therapy.1,58,59 Useful acronym to follow the patient from tumor diagnosis to potential checkpoint inhibitors related myocardial injury. ICI = immune checkpoint inhibitors.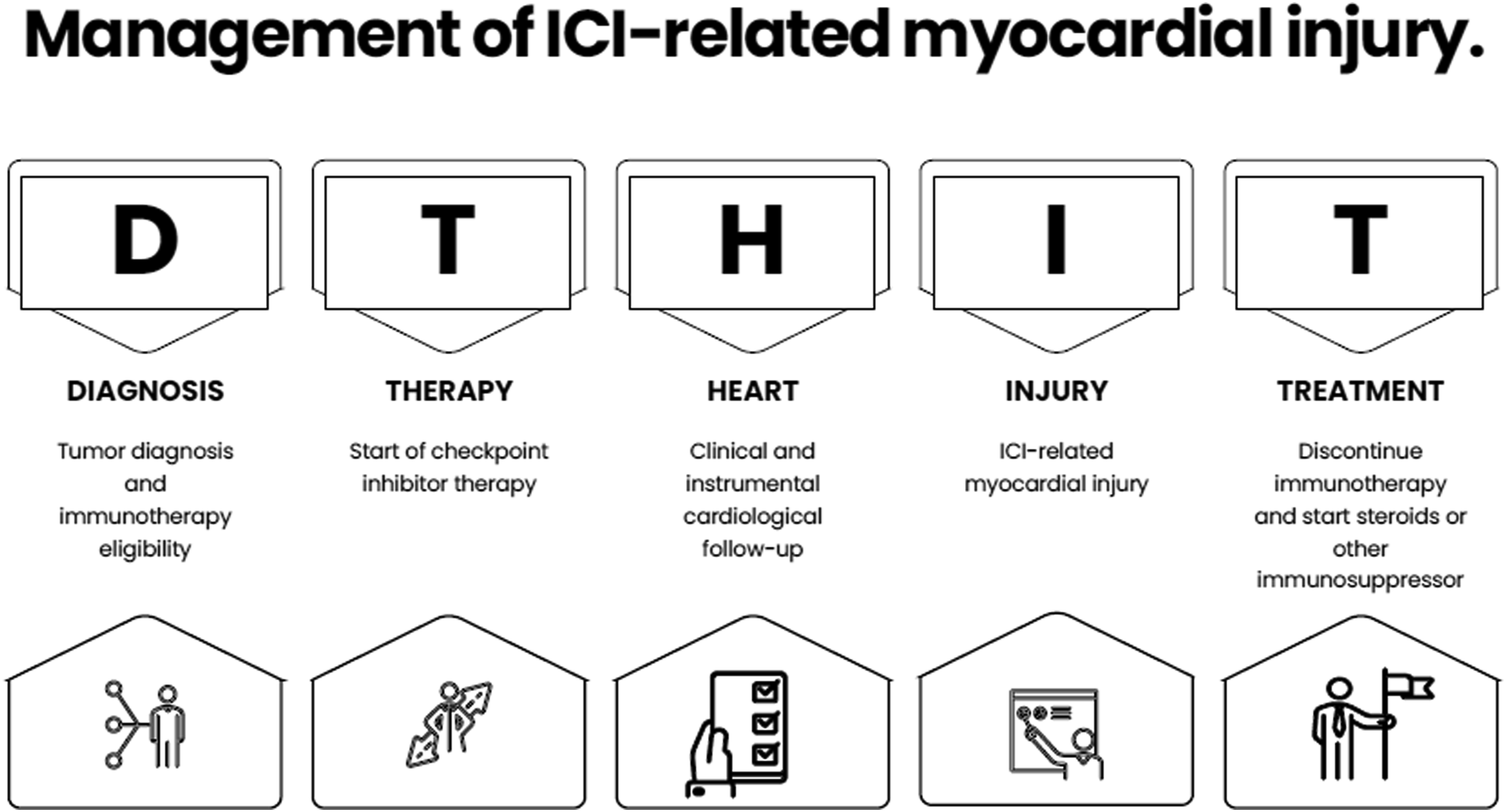
Major cases of immuno-related myocardial injury: symptoms, treatment, and prognosis.
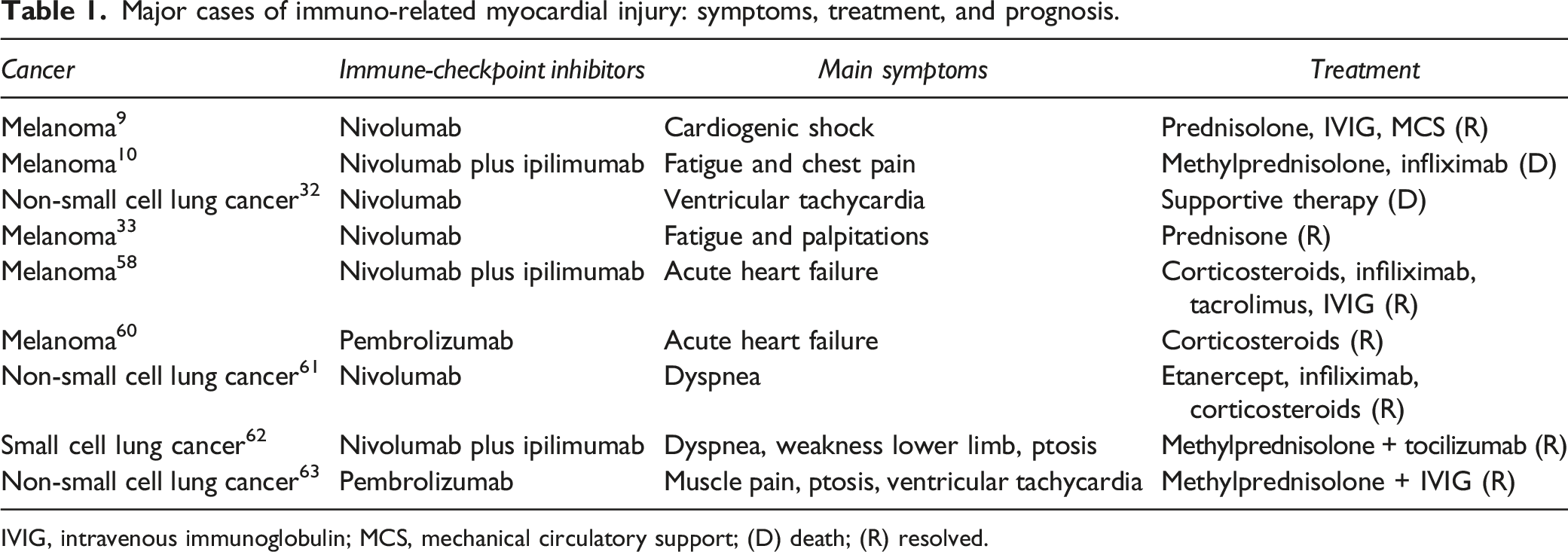
IVIG, intravenous immunoglobulin; MCS, mechanical circulatory support; (D) death; (R) resolved.
Main classes of drugs used for the treatment of immuno-related myocarditis.
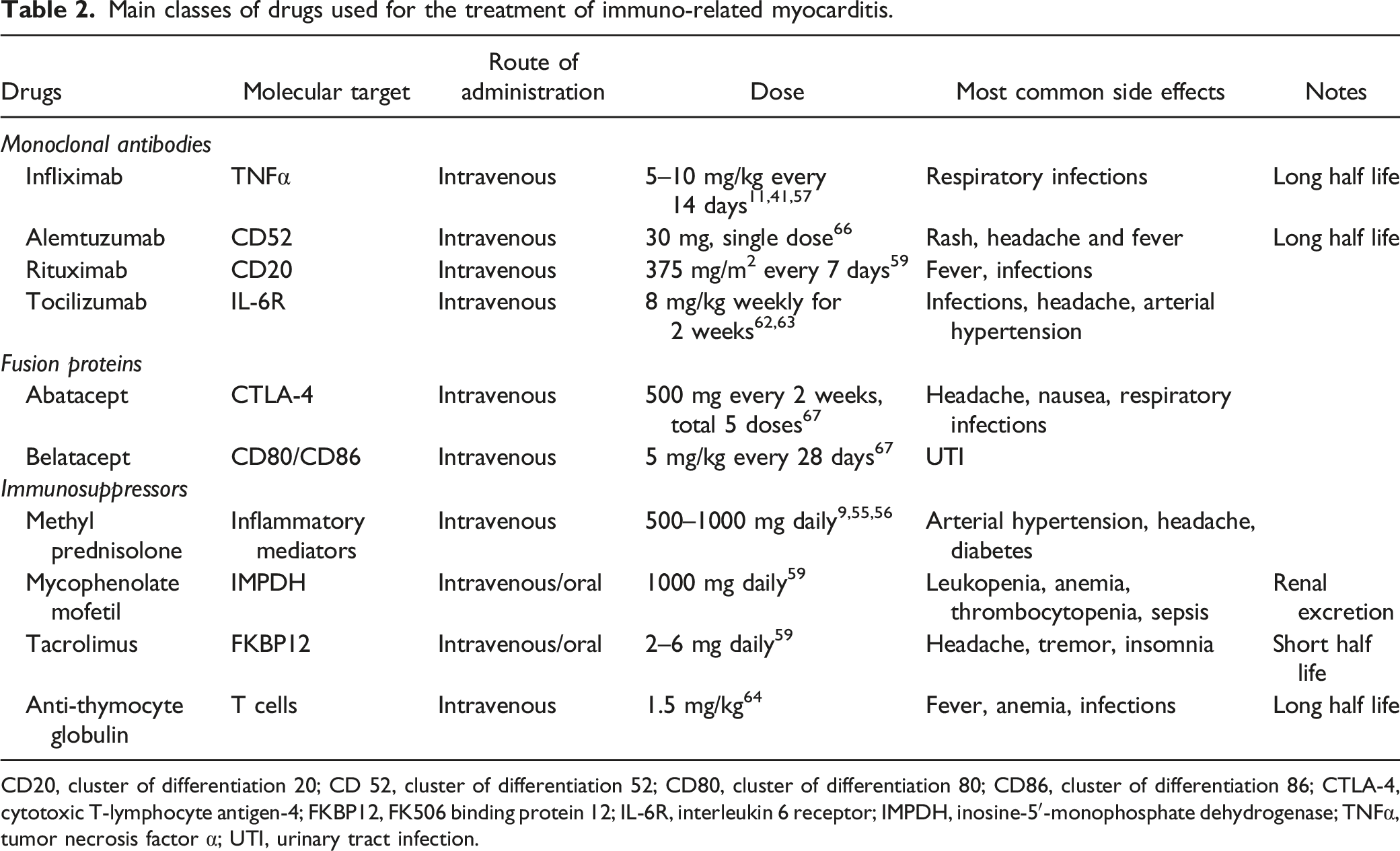
CD20, cluster of differentiation 20; CD 52, cluster of differentiation 52; CD80, cluster of differentiation 80; CD86, cluster of differentiation 86; CTLA-4, cytotoxic T-lymphocyte antigen-4; FKBP12, FK506 binding protein 12; IL-6R, interleukin 6 receptor; IMPDH, inosine-5′-monophosphate dehydrogenase; TNFα, tumor necrosis factor α; UTI, urinary tract infection.
Patients with life-threatening arrhythmias, a common complication of ICI-induced myocarditis, should receive appropriate anti-arrhythmic agents such as intravenous amiodarone and be considered for temporary or potentially permanent pacemaker placement for advanced conduction disease. Classical drugs used in heart failure management such as angiotensin converting enzyme (ACE) inhibitors and β-blockers are suggested, particularly in the case of LV dysfunction (LVEF <50%), and high-dose aspirin could be required as auxiliary therapies for patients with heart failure and raised troponin in the context of cardiac ischemia.1,50 In case of sick sinus syndrome, a low dose of CS (12.5 mg/day) taken orally a day might control symptoms. 69 For persistent refractory heart failure, in addition to vasoactive drugs, intra-aortic balloon pump (IABP), ventricular support devices, or extracorporeal membrane oxygenation (ECMO) support might be attempted to support critically ill patients.70–74
In case of pericardial disease, in addition, to interrupt ICI administration, current practice is to administer colchicine and non-steroidal anti-inflammatory drugs (NSAIDs), following the guidelines. 75 Pericardiocentesis and hemodynamic support have to be considered if cardiac tamponade is present. 75
Where will we go?
Targeted therapies and ICIs have greatly improved cancer overall survival, even though cardiac adverse effects remain a critical diagnosis and management issue.
In order to optimize targeted therapy sensitivity and specificity for diagnosis, in an experimental preclinical autoimmune myocarditis model (EAM), non-invasive contrast-enhanced ultrasound molecular imaging using microbubbles targeted to CD4+ lymphocytes and P-selectin endothelial cell adhesion molecule has shown utility in improving diagnosis and prediction of LV fibrosis. 76 However, this approach is still far from being clinically applied.
The near future is imaging with radionuclides which is currently used for the therapeutic follow-up of tumors; in the future, for example, 18F-FDG PET/CT could be useful for the early identification of adverse reactions, combining anatomical data with functional data. 77 MicroRNAs regulate many immune processes, including function, homeostasis and phenotypic stability of T-reg cells, inhibiting dendritic cells (DC) activation and blocking Th1/Th17 immune responses. MiR-223-3p plays an important role in cancer development as a tumor suppressor gene, blocking cell proliferation and migration, regulating epithelial-mesenchymal transition and protecting against the development of autoimmune myocarditis as highlighted in murine models, showing itself as an interesting future cardioprotective target.78,79
Myocardial inflammation may lead to contractile dysfunction, provoking hemodynamic compromise and an increase in wall stress: mechanical stress induces a pro-fibrogenic transition of T-lymphocytes and macrophages, therefore local and systemic activation of the renin-angiotensin-aldosterone system in overloaded hearts contributes to inflammation and myocardial remodeling. 80 Mechanical unloading of LV through axial flow pumps devices may reduce myocardial work, oxygen demand and consequently wall stress and lymphocyte activation.80,81 In order to control myocardial inflammation, Watanabe et al. have recently pointed out that linagliptin, a xanthine-based dipeptidyl peptidase-4 inhibitor, could improve EAM and ICI-induced myocarditis, interacting with cathepsin G and suppressing oxidative stress, thus reducing lymphocytic-macrophages infiltration, myocardial damage, and fibrosis. 82
Environmental factors are considered critical in autoimmunity pathogenesis. Gut microbiota could prime T-helper cells against bacterial antigens that mimic cardiac proteins, like myosin heavy chain. 83 Subclinical ischemia or inflammation induced by ICI could allow antigen exposure, cross-reactive microbiota-driven T-helper cells may promote myocardial damage, so broad-spectrum antibiotic treatment could dampen T-helper cell-mediated inflammatory responses.83,84
A recent cellular and preclinical model evidenced that the mechanism of cardiotoxicity could be driven by NOD-, LRR-, and pyrin domain-containing protein 3/Myeloid differentiation primary response 88 (NLRP3/MyD88) pathways and pro-inflammatory cytokine storms in myocardial tissue. Thus, specific inhibitors may block this cascade. 85 The most interesting target seems to be interleukin-6 (IL-6), a critical driver of acute and chronic inflammation, whose signaling promotes a pro-tumorigenic immune-suppressive network. 86 Therefore, blocking the IL-6 pathway could carry complementary anti-tumor properties and improve the differentiation of CD4+ T cells into gamma Interferon (IFN-γ), producing effector T-helper type 1 cells. 87 Furthermore, accumulating evidence suggest that the IL-6-Th17 pathway may have an important role in the pathogenesis of immune-related adverse events, especially in steroid-refractory cases.88,89 IL-17A-expressing CD4+ T cells [c-Kit− CD161+ MDR (multi drug resistance)1 + Th17 cells] have been reported as key effectors of autoimmune inflammation refractory to glucocorticoids. 87 The pathogenic effect of IL-6 is essential in the differentiation of pro-inflammatory Th-17 cells from naïve CD4+ T cells, which might suggest a role for this Th17 subset in steroid-refractory immune-related adverse events. 90 Tocilizumab (TCZ), 8 mg/kg body weight weekly for two doses, is used to treat severe or life-threatening chimeric antigen receptor (CAR) T cell-induced cytokine release syndrome (CRS) in adults and in pediatric patients. 62 Compared with the other available selective immune-suppressive drugs, the anti-IL-6R agent TCZ offers several strategic advantages. It appears to be safe for the CV system, with a low complication rate and without the risk of compromising ICIs efficacy.62,63
Immunosuppressive therapy still represents the mainstay of treatment of drug-induced cardiac injury, although we have a lack evidence on which drug use as first-line therapy and the duration of therapy. Generally, therapy involves a CS, and if there is no response, a non-steroidal immunosuppressant is switched, either alone or in combination. 91 Every regimen has to be evaluated against the risk of infection, including reactivation of latent cardiac microbial infections, and cancer response. The dose of CS required to produce anti-inflammatory action is lower, compared with the immunosuppressive dose. Combining prednisolone with potent immunosuppressant agents could allow steroid dose reduction and preserve anti-inflammatory effects, in order to shield patients against high-dose steroid effects, especially metabolic and arrhythmogenic ones.
It is mandated to start treatment at the diagnosis, with high dosages of methylprednisolone between 500 and 1000 mg in order to detect early steroid-resistant forms. The duration of CS therapy should be no less than 4 weeks and continued despite the normalization of clinical and laboratory parameters. CS is not always sufficient to obtain a complete resolution of the condition. Thus, continuous multiparametric monitoring is essential to identify resistant forms in order to switch to a different immunosuppressive regimen. In case of therapeutic failure, monitoring disease course with Tn-I and pro-BNP, it is recommended to use a second-line treatment with tocilizumab, which, however, should be preferred as first-line treatment in the suspicion of aggressive myocarditis. In case of anti-CTLA 4 toxicity, the first choice should be a CTLA-4 agonist such as abatacept and belatacept, if available. The combination of abatacept and ruxolitinib (Janus kinase 1 and 2 inhibitor) could be a treatment option for ICI-related myocarditis; this hypothesis requires further future evaluation of the safety profile. 92 In view of current pathogenic hypotheses, it is always advisable to associate plasmapheresis to the immunosuppressive treatment in order to reduce the autoantibody load. Supportive cardiological therapy is also essential, considered the higher risk of MACE, in order to give the time for immunosuppressive treatment to be effective.
Conclusion
Future challenges will be improving therapeutic targets, increasing early detection of cardiac damage, and developing optimal duration of treatment. These goals could only be reached with the multidisciplinary efforts of oncologists, cardiologists, radiologists, immunologists, and emergency physicians, prioritizing the proper treatment for each patient. Finally, the identification of genetic determinants of immune-related cardiotoxicity represents another future perspective.
Footnotes
Author Contributions
All authors contributed to: (1) substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data, (2) drafting the article or revising it critically for important intellectual content, and (3) final approval of the version to be published.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.