
Abstract
This study assessed the role of nucleotide-binding domain and leucine-rich repeat containing receptor, caspase recruitment domain containing 5 (NLRC5) in macrophages in atherosclerotic plaque formation in acute coronary syndromes (ACS) by modulating the nuclear factor-kappaB (NF-κB) cascade. Peripheral blood was obtained from ACS patients and matched controls, and NLRC5 expression and DNA methylation were analyzed. In vitro, peripheral blood mononuclear cells from donors were induced into macrophage-derived foam cells and transfected with small interfering RNA negative control (si-NC) or si-NLRC5 plasmids to assess foam cell formation and cytokine release. In vivo, ApoE−/− mice fed a high-fat diet and subjected to NLRC5 silencing were used as an ACS model. Peritoneal macrophage phagocytosis, aortic lipid accumulation, plaque size, and collagen fiber content were evaluated, while lipid metabolism- and inflammation-related genes were measured in foam cells and aortas. NLRC5 was highly expressed and hypomethylated in ACS patients. NLRC5 knockdown suppressed foam cell formation and inflammation in vitro. In ACS mice, silencing NLRC5 reduced lipid levels and cytokines, inhibited lipid deposition, decreased plaque size, and enhanced collagen fiber content through NF-κB pathway inhibition. These findings suggest that NLRC5 silencing may protect against atherosclerosis in ACS by regulating macrophage function and inflammatory signaling.
Keywords
Introduction
Acute coronary syndrome (ACS) encompasses a spectrum of coronary artery disorders, including unstable angina, ST-segment elevation myocardial infarction, and non-ST segment elevation myocardial infarction. 1 The primary pathological basis of ACS is the rupture or erosion of atherosclerotic plaques, which subsequently triggers thrombus formation—although non-atherosclerotic mechanisms may also contribute in some cases. 2 The concept of the “vulnerable plaque” has become central to understanding ACS pathogenesis. These plaques are typically characterized by a large lipid core, abundant macrophage-derived foam cells, and cellular debris resulting from apoptosis and necrosis. 3 Notably, macrophages play a pivotal role in atherosclerosis (AS) by sustaining local inflammation, promoting plaque progression, and facilitating thrombotic events. 4 Therefore, targeting macrophage-driven plaque development represents a promising therapeutic strategy in ACS management.
Among the innate immune components involved in these processes, nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins serve as intracellular sensors that detect microbial and endogenous danger signals, mediating inflammatory responses and regulated cell death. 5 Within this family, nucleotide-binding domain and leucine-rich repeat containing receptor, caspase recruitment domain containing 5 (NLRC5)—the most structurally extensive NLR member—has garnered increasing attention. NLRC5 has been shown to influence atherosclerosis in ACS through the modulation of macrophage signal transducer and activator of transcription 3 (STAT3) signaling. 6 In other studies, NLRC5 upregulation by ubiquitin-specific protease 14 was found to inhibit endothelial cell activation and inflammatory responses in AS. 7 Additionally, NLRC5 deficiency alleviated cardiac fibrosis in diabetic cardiomyopathy by inhibiting endothelial-mesenchymal transition via the Sma and Mad related proteins (Smad)2/3 signaling cascade. 8
Inflammation plays a central role in thrombotic disease progression, with nuclear factor-kappaB (NF-κB) serving as a key regulator of pro-inflammatory and pro-coagulant responses.9,10 For example, interleukin (IL)-34-mediated NF-κB activation promotes C-C motif chemokine ligand 2 (CCL2) expression, enhances macrophage recruitment and polarization, and contributes to adverse cardiac remodeling and heart failure following ischemia-reperfusion injury, 11 highlighting the critical role of the NF-κB pathway in ACS. Furthermore, interactions between NLRC5 and NF-κB signaling have been reported in various cell types.12 -14 Interestingly, NLRC5 has been described as a negative regulator of NF-κB-mediated inflammatory signaling. 15
Despite these insights, the precise role of NLRC5 in macrophage-mediated NF-κB signaling during atherosclerotic plaque formation in ACS remains unclear. Therefore, this study aims to explore the functional relationship between NLRC5 and the NF-κB pathway in macrophages and its impact on plaque development in the context of ACS.
Materials and Methods
Ethical Approval
All patients provided written informed consent prior to participation. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of The Second Affiliated Hospital of Kunming Medical University. All animal experiments were performed in accordance with institutional ethical guidelines and were approved by the Animal Ethics Committee of The Second Affiliated Hospital of Kunming Medical University.
Patients and Controls
Sixty patients diagnosed with ACS and 60 age- and sex-matched controls were recruited from The Second Affiliated Hospital of Kunming Medical University between May 2022 and April 2024. ACS cases included individuals with unstable angina and acute myocardial infarction. All patients presented with ST-segment changes and/or T-wave inversions, and the diagnosis of myocardial infarction was confirmed by significantly elevated levels of creatine kinase-MB (CK-MB) and cardiac troponin I. The study population also included patients with non-ST-segment elevation myocardial infarction and confirmed coronary artery disease. Exclusion criteria included any history of coronary artery bypass grafting, cardiogenic shock, stroke, malignancy, or acute/chronic infections. Controls had no known diagnoses of hypertension, hyperlipidemia, or cardiovascular diseases. Informed consent was obtained from all participants, and the study protocol was approved by the institutional ethics committee.
Bisulfite Pyrophosphate Sequencing
Genomic DNA was extracted from peripheral blood samples following magnetic bead sorting. Target regions for NLRC5 methylation analysis (cg16411857 and cg07839457 loci) were selected using PyroMark Assay Design Software (Qiagen, Hilden, Germany). Bisulfite conversion was performed, and PCR amplification was carried out using hot-start Taq DNA polymerase (Qiagen). The resulting amplicons were analyzed using a PyroMark Q24 pyrosequencer (Qiagen), and methylation levels were calculated as the ratio of cytosine (C) to total cytosine plus thymine (C + T) using PyroMark Q24 software v2.0.8. 16
Macrophage Extraction and Induction
Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll density gradient centrifugation and resuspended in phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA). Monocytes were then differentiated into macrophages by culturing with 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) 17 in 6-well plates at 10 × 106 cells/well. To induce foam cell formation, macrophages were incubated overnight with 50 μg/mL acetylated low-density lipoprotein (acLDL; KyvoBio, Shanghai, China). After induction, cells were fixed in 4% paraformaldehyde for 30 minutes and washed with precooled 60% isopropanol. Oil Red O staining (Sigma-Aldrich, Shanghai, China; cat no 1320-06-5) was performed at room temperature for 30 minutes to assess lipid accumulation. Cells were washed, resuspended in 200 μL PBS, and observed under a fluorescence inverted microscope (KEYENCE, Shanghai, China) at 600× magnification.18,19
Cell Transfection
Foam cells were cultured in 96-well plates with Dulbecco’s modified Eagle medium (DMEM; Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) at 37 °C and 5% CO2. Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 48 hours. Cells were transfected with either small interfering RNA (si)-NLRC5 or si-NC plasmids. si-NLRC5 F: 5′-CCGGAATTCCGGGATGGCCAGAAGCTGGA-3′, R: 5′-GGGACCCCCGTCACCTGAGTCTTCCCA-3′, si-NC F: 5′-UUCUCCGAACGUGUCACGUTT-3′, R: 5′-ACGUGACACGUUCGGAGAATT-3′. 20
Animal Model and Tissue Collection
This study employed specific pathogen-free (SPF) grade ApoE−/− mice (6 weeks old, 18-20 g), and NLRC5 knockout (NLRC5−/−) mice derived from the ApoE−/− background, obtained from Shanghai Biomodel Organism (Shanghai, China). 21 All mice were divided into groups (n = 6/group): control group (ApoE−/− mice fed a standard diet), ACS group (ApoE−/− mice with ACS modeling), dehydroxymethylepoxyquinomicin (DHMEQ) group (ACS-induced ApoE−/− mice treated with the NF-κB inhibitor DHMEQ), NLRC5−/− group (ACS-induced NLRC5−/− mice), and NLRC5−/− + DHMEQ group (ACS-induced NLRC5−/− mice treated with DHMEQ). All mice were housed under SPF conditions (19 °C-22 °C, 50%-70% humidity, adequate ventilation). Except for the control group, mice were fed a high-fat diet containing 15% fat and 1.25% cholesterol. The NF-κB inhibitor DHMEQ (HY-14645; MCE, Shanghai, China) was administered via intraperitoneal injection at 10 mg/kg, 3 times/week. 22 Blood samples were collected from the orbital venous plexus at 10 and 12 weeks for the assessment of lipid profiles and inflammatory markers. After euthanasia by cervical dislocation at week 12, aortic root tissues were harvested for histological examination. Successful model establishment was confirmed by Oil Red O staining and evident plaque formation in the aorta.
Lipid Profile Measurement
At 12 weeks post-treatment, mice were fasted for 8 hours prior to sample collection. Approximately 1 mL of blood was obtained from the tail vein, centrifuged to isolate serum, and stored at −80 °C until analysis. Commercial assay kits were used to quantify: total cholesterol (TC; 60723ES60; Yeasen, Shanghai, China), triglycerides (TG; MAK564; Sigma, Shanghai, China), high-density lipoprotein cholesterol (HDL-C; 60737ES; Yeasen), and low-density lipoprotein cholesterol (LDL-C; 60736ES; Yeasen).
Extraction and Characterization of Mouse Peritoneal Macrophages
Twelve weeks after treatment, mice were intraperitoneally injected with 1.0 mL of 4% thioglycollate broth. After 72 hours, peritoneal cells were harvested under sterile conditions, centrifuged, and resuspended in RPMI-1640 medium. Cell suspensions were seeded into 24-well plates and incubated for 4 hours to allow macrophage adherence. Following medium change, cells were maintained at 37 °C in a 5% CO2 incubator for 7 days. 23
Ink Phagocytosis Assay
To assess phagocytic capacity, 20 μL of 10% diluted India ink was added to peritoneal macrophage cultures and incubated for 3 hours. Cells were then washed 3 times with RPMI-1640, fixed in 10% formaldehyde, and mounted with glycerol gelatin. Under 600× magnification (Olympus, Tokyo, Japan), macrophages containing visible black ink particles were counted as phagocytosis-positive cells. 23
Enzyme-Linked Immunosorbent Assay (ELISA)
Levels of inflammatory cytokines were quantified using enzyme-linked immunosorbent assay kits according to the manufacturers’ protocols. The following kits were used: IL-6 (cat no 88-7064-88; Invitrogen, Carlsbad, CA, USA), IL-1β (BMS6002-2TEN; Invitrogen), tumor necrosis factor-α (TNF-α; BMS607-3TEN; Invitrogen), monocyte chemoattractant protein-1 (MCP-1; BMS6005TEN; Invitrogen), and vascular cell adhesion molecule-1 (VCAM-1; EMVCAM1; Invitrogen).24,25
Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from cells, tissues, and blood samples using TRIzol reagent (Tiangen, Beijing, China). RNA purity and concentration were determined spectrophotometrically at 260/280 nm (Biotek, Winooski, VT, USA). A maximum of 2000 ng RNA was reverse-transcribed into cDNA using the RT SuperMix kit (Tiangen, China). Quantitative PCR was performed using SYBR Green chemistry. Relative gene expression was normalized to β-actin and calculated using the 2−ΔΔCt method. Primer sequences are listed in Table 1.26,27
NLRC5 Primer Sequences.
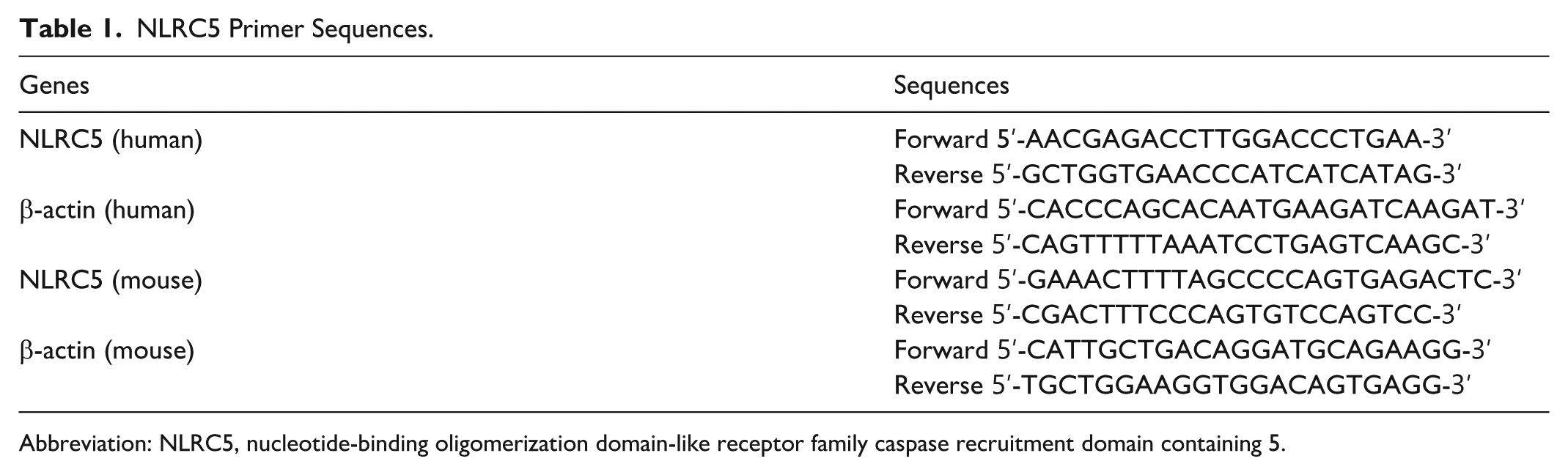
Abbreviation: NLRC5, nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 5.
Western Blot
Proteins were extracted from cells and tissues using RIPA lysis buffer (S7705; Sigma, Shanghai, China), and concentrations were determined by BCA assay. Equal amounts of protein were separated by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (IPFL00010; Sigma). Membranes were blocked with 5% bovine serum albumin for 1 hour at 37 °C and incubated overnight at 4 °C with primary antibodies: NLRC5 (ab117624; Abcam, Cambridge, MA, USA, 1:500), p-IκB (Cell Signaling Technology, Danvers, MA, USA; 9246, 1:1000), p-p65 (AF2006; Affinity, Changzhou, China; 1:1000), and β-actin (Sigma-Aldrich, St Louis, MO, USA; A5441, 1:5000). After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (S0001; Affinity; 1:1000) for 1 hour. Signals were detected using an enhanced chemiluminescence (ECL) reagent (TANON, China), and band intensity was visualized with an Amersham Imager 600 (GE Healthcare Life Sciences, Chicago, IL, USA). Densitometric analysis was performed using ImageJ software v1.8.0 (National Institutes of Health, Bethesda, MD, USA). 28
Hematoxylin-Eosin (HE) Staining
Frozen sections of mouse aortic tissue were stained with hematoxylin and washed with tap water. The slides were then differentiated with 1% hydrochloric acid ethanol, counterstained with 0.5% eosin for 2 minutes, dehydrated sequentially in 85%, 95%, and absolute ethanol (I and II), cleared in xylene (I and II), and mounted. Morphological assessment was performed under a 200× microscope (Olympus, Tokyo, Japan), and images were analyzed using ImagePro Plus 6.0 (Media Cybernetics, Rockville, MD, USA). Five random fields were selected per section, and the plaque area/cross-sectional area ratio of the arterial wall was calculated. 23
Masson Staining
Frozen sections were stained with hematoxylin and differentiated in 1% acid ethanol for 3 minutes. Slides were then stained with Ponceau S for 10 minutes, rinsed in 0.2% acetic acid for 5 minutes, and differentiated with phosphomolybdic acid solution for 5 minutes. Sections were subsequently stained with aniline blue for 5 minutes, dehydrated, cleared, and mounted. Images were obtained under a 200× microscope. Collagen fibers appeared blue, and muscle tissue red. Five random fields were analyzed per section, and the collagen area/vascular area ratio was calculated using ImagePro Plus 6.0 (Media Cybernetics, Rockville, MD, USA). 23
Oil Red O Staining
Frozen aortic tissue sections were fixed and rinsed with 60% isopropanol, followed by staining with Oil Red O solution. After staining, the sections were briefly washed and differentiated with 1% hydrochloric acid. The stained sections were then mounted using glycerol gelatin and examined under a light microscope at 200× magnification. Quantitative analysis of lipid deposition was performed using ImagePro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA). 23
Statistical Analysis
All data analyses were performed using GraphPad Prism 8.0.1 (GraphPad Software, Inc, San Diego, CA, USA). All experiments were conducted in triplicate unless otherwise specified. Quantitative data are presented as mean ± standard deviation (SD). Two-group comparisons were analyzed using unpaired Student’s t-test. Multiple group comparisons were performed using 1-way analysis of variance (ANOVA) followed by appropriate post hoc tests. Categorical data were expressed as percentages and compared using the χ2 (chi-square) test. Statistical significance was defined as: *P < .05, **P < .01, ***P < .001 in 2-sided tests. 29
Results
NLRC5 Is Upregulated in ACS and Associated With Disease Progression
As shown in Table 2, significant differences in high-sensitivity C-reactive protein and CK-MB levels were observed between the ACS and control subjects, while other clinical parameters showed no significant differences. In ACS patients, NLRC5 gene expression in peripheral blood was approximately twice the level found in controls (Figure 1A). NLRC5 locus methylation was significantly reduced in blood samples from ACS patients compared with controls (Figure 1B), indicating that NLRC5 upregulation may be epigenetically regulated via hypomethylation. Peripheral blood mononuclear cells (PBMCs) from controls were successfully induced into foam cells by acLDL stimulation, and Oil Red O staining confirmed substantial lipid accumulation in the cytoplasm (Figure 1C). RT-qPCR assay further confirmed elevated NLRC5 expression in foam cells compared with unstimulated macrophages (Figure 1D), indicating a potential role for NLRC5 in promoting foam cell formation and the progression of ACS.
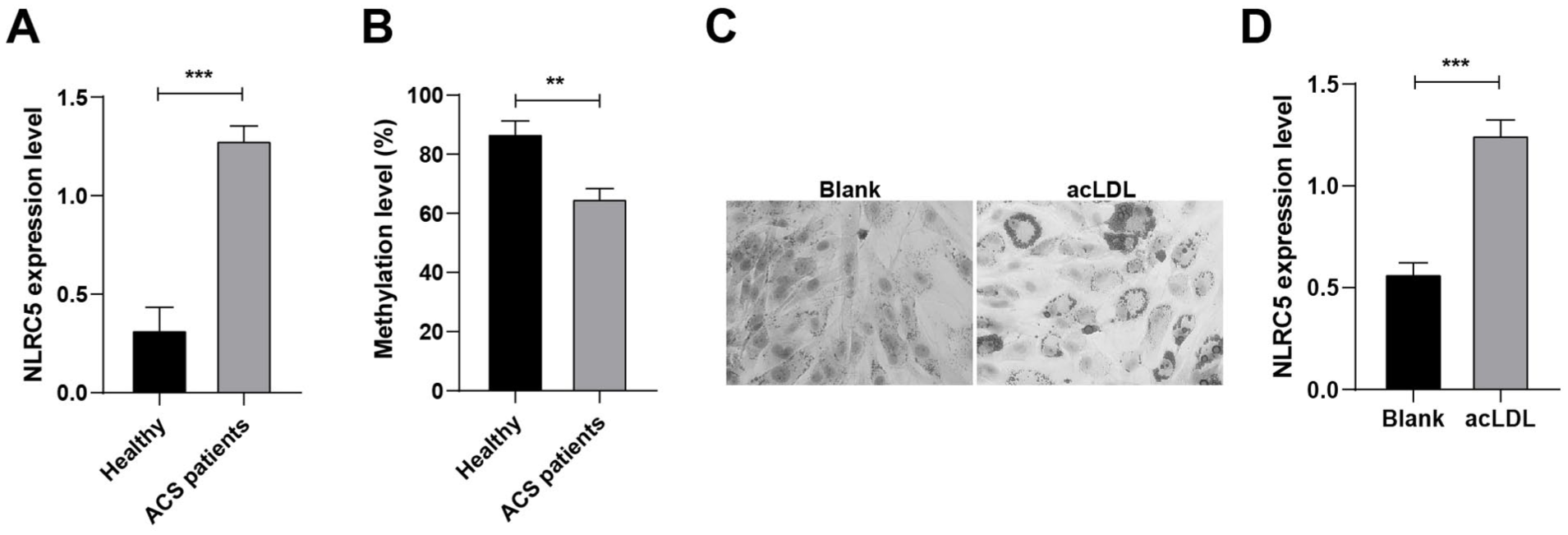
NLRC5 is highly expressed in ACS and promotes disease progression. (A) RT-qPCR to determine the expression level of NLRC5 (n = 60). (B) Bisulfite pyrophosphate to determine the methylation level of NLRC5 (n = 60). (C): Oil Red O staining to detect foam macrophage formation (×600). (D) RT-qPCR to determine NLRC5 expression levels in foam macrophages. **P < .01, ***P < .001.
Comparison of Clinical Characteristics Between ACS Patients and Controls.
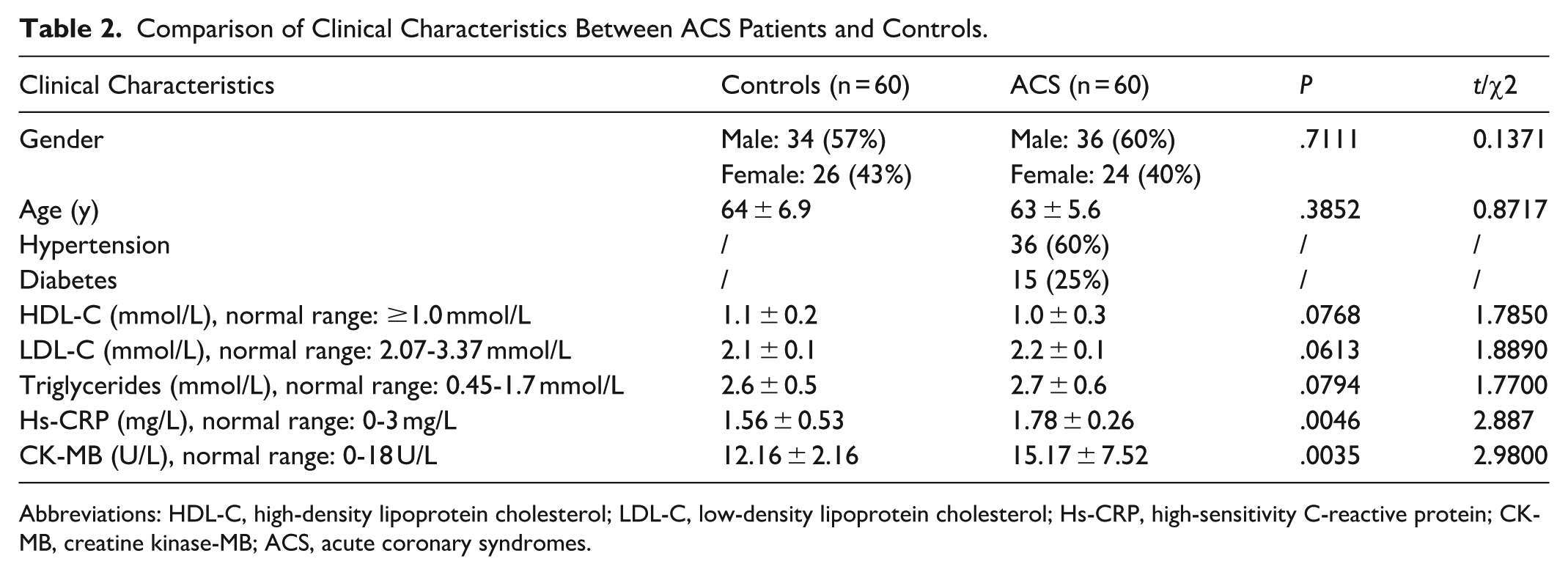
Abbreviations: HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Hs-CRP, high-sensitivity C-reactive protein; CK-MB, creatine kinase-MB; ACS, acute coronary syndromes.
NLRC5 Knockdown Inhibits Inflammatory Response of Foam Cells
si-NC or si-NLRC5 plasmid was successfully transfected into PBMC-derived foam cells. NLRC5 expression in foam cells was significantly reduced after transfection with si-NLRC5, confirming effective knockdown (Figure 2A). Reduction of NLRC5 lowered TNF-α, IL-1β, and IL-6 (Figure 2B), indicating that NLRC5 knockdown suppressed the inflammatory capacity of foam cells. Oil Red O staining revealed abundant intracellular lipid deposition in the acLDL and si-NC groups, while lipid accumulation was notably reduced in the si-NLRC5 group (Figure 2C), reflecting that knocking down NLRC5 reduced foam cell formation.
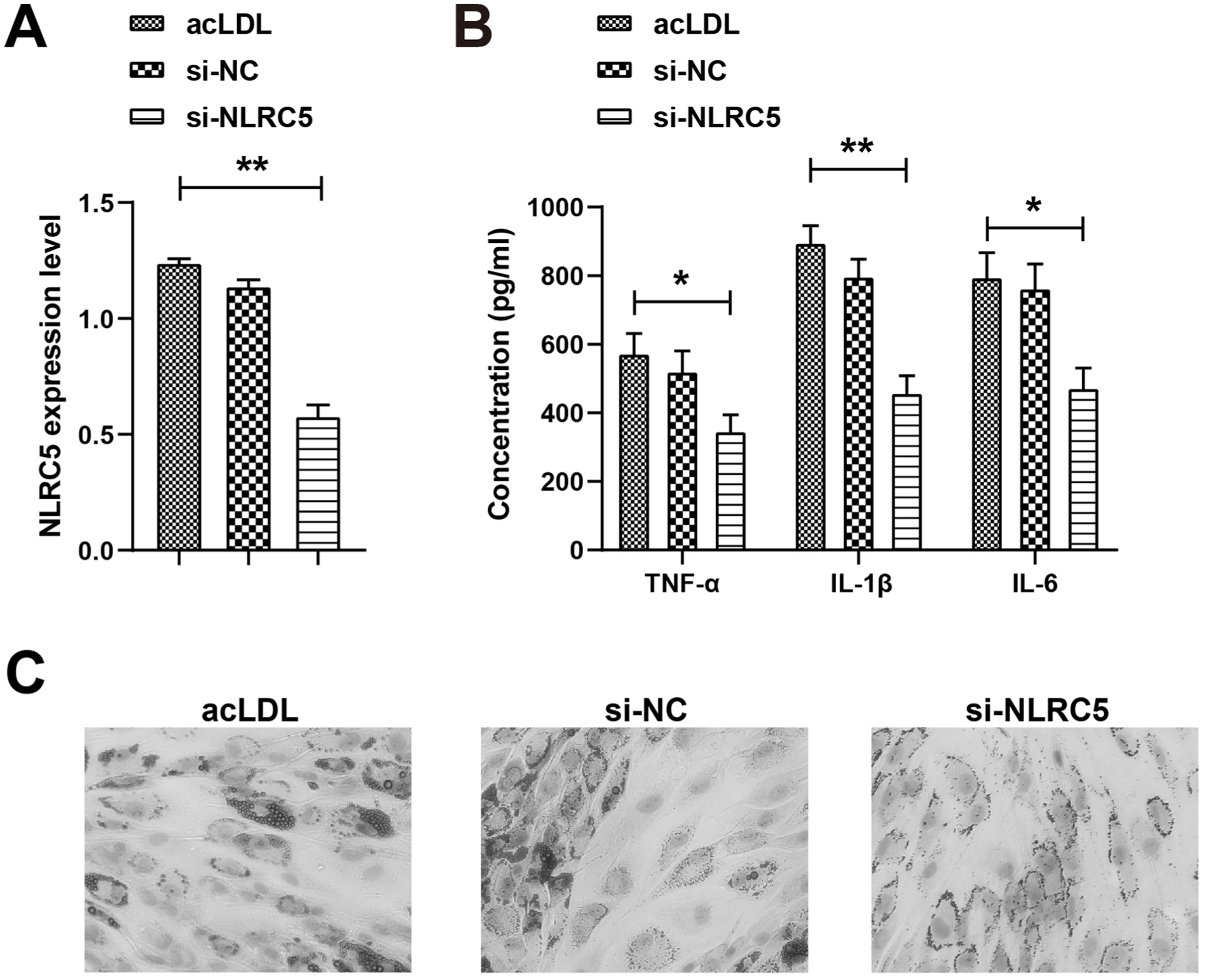
NLRC5 knockdown inhibits the inflammatory response of foam cells. (A) RT-qPCR to detect the expression level of NLRC5. (B) ELISA to determine the levels of inflammatory factors TNF-α, IL-1β, and IL-6. (C) Oil Red O staining to detect foam cell formation (×600). *P < .05, **P < .01.
NLRC5 Knockdown Reduces Lipid Levels and Inflammation in ACS Mice
An ApoE−/− mouse model of ACS fed a high-fat diet was successfully created to study the in vivo role of NLRC5. Increased NLRC5 levels were found in the aortas of ACS mice (Figure 3A). TG, TC, and LDL-C levels were significantly raised, whereas HDL-C levels were significantly reduced in ACS mice (Figure 3B–E). These lipid abnormalities were significantly alleviated by NLRC5 knockdown. NLRC5−/− mice had significantly reduced concentrations of TNF-α, IL-1β, and IL-6, and reduced levels of vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1; Figure 3F). Collectively, these results indicate that NLRC5 knockdown alleviates both lipid accumulation and inflammatory responses in ACS mice.
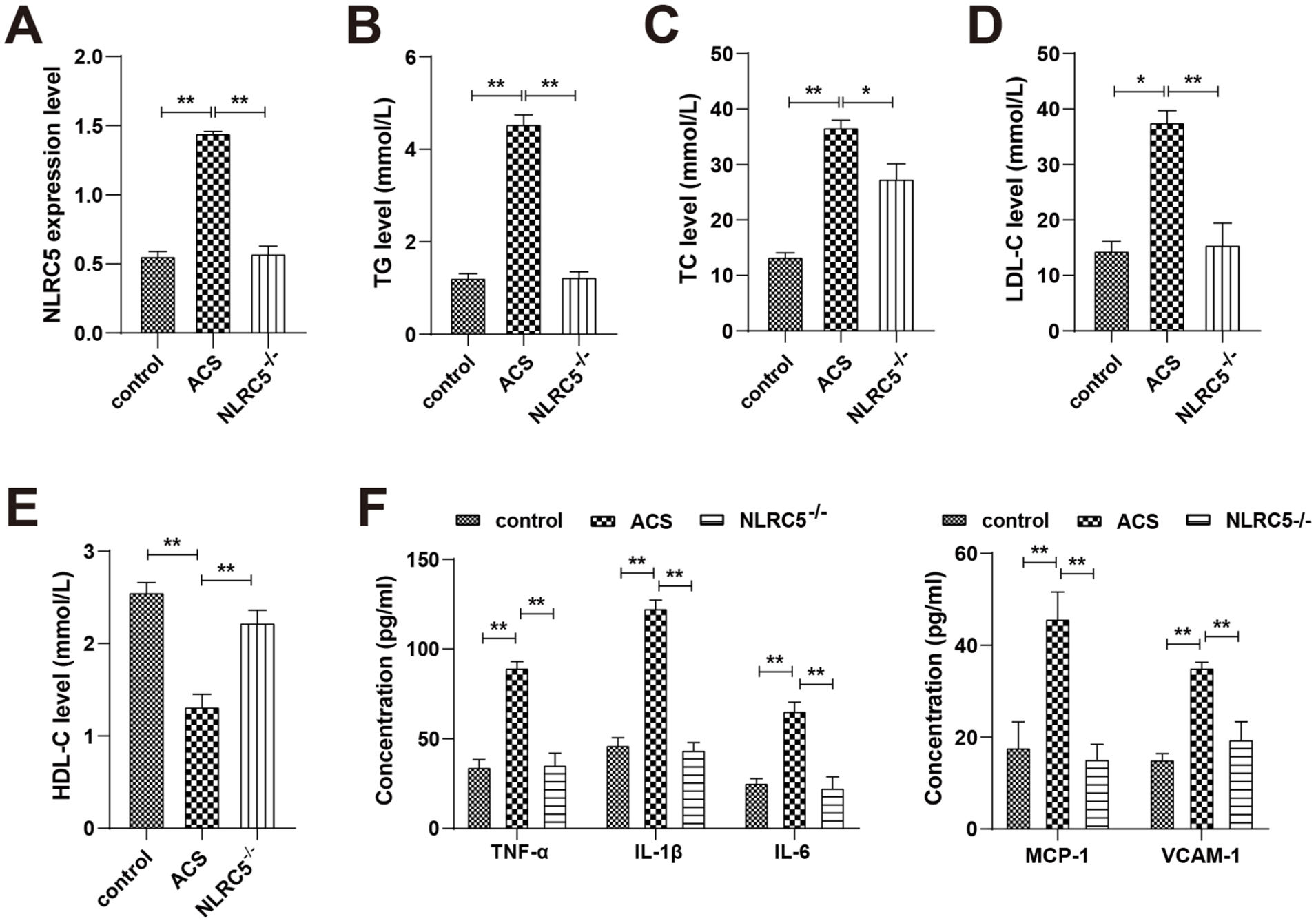
NLRC5 knockdown reduces lipid and inflammation levels in ACS mice. (A) RT-qPCR to detect NLRC5 expression. (B–E) Detection of lipid levels (TC, TG, HDL-C, and LDL-C). (F) ELISA to determine levels of TNF-α, IL-1β, IL-6, VCAM-1, and MCP-1. *P < .05, **P < .01.
NLRC5 Knockdown Inhibits Activation of the NF-κB Cascade
As reported, NLRC5 influences the secretion of pro-inflammatory cytokines via the NF-κB or inflammasome pathways. 28 Western blot results showed that NLRC5 knockdown inhibited the phosphorylation of p65 and IκB in PBMC-derived foam cells, and decreased p-p65 and p-IκB protein expression levels (Figure 4A), indicating inhibition of NF-κB activation. Further in vivo experiments showed (Figure 4B–D) that NLRC5−/− ACS mice and those treated with DHMEQ ghad similarly reduced NLRC5 expression and suppression of p-p65 and p-IκB protein levels, both significantly lower than those in the ACS group. Interestingly, NF-κB inhibition was more pronounced in the NLRC5−/− group alone compared to the combination of NLRC5 knockdown and DHMEQ treatment, suggesting a possible synergistic inhibitory effect of genetic suppression of NLRC5 and pharmacological NF-κB inhibition.
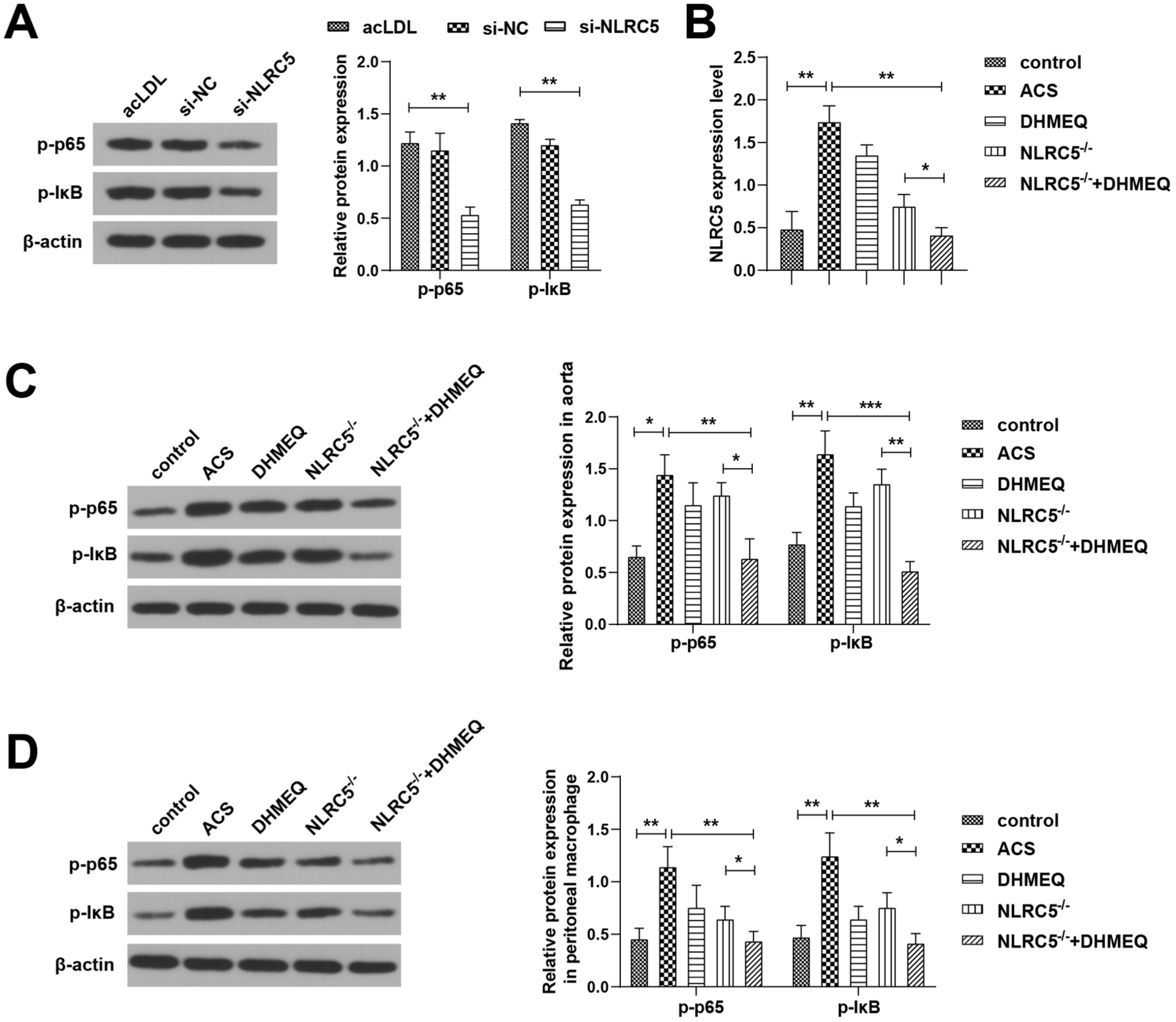
NLRC5 knockdown inhibits the activation of the NF-κB cascade in cells and ACS mice. (A) Western blot to determine the levels of p-p65 and p-IκB in foam macrophages. (B) RT-qPCR to detect the expression levels of NLRC5 in peritoneal macrophages of ACS mice. (C) Western blot to detect p-p65 and p-IκB in mouse aorta. (D) Western blot to detect p-p65 and p-IκB in peritoneal macrophages of ACS mice. *P < .05, **P < .01, ***P < .001.
NLRC5 Knockdown Inhibits Atherosclerotic Plaque Formation in ACS by Regulating the NF-κB Cascade
To further investigate whether NLRC5 mediates ACS progression through regulation of the NF-κB signaling pathway, histological staining was performed on aortic tissues from ACS model mice, and peritoneal macrophages isolated from these mice were used to assess phagocytic function. Phagocytosis assays revealed that NLRC5 knockdown significantly inhibited macrophage phagocytic activity, resulting in a reduced number of ink-positive cells (Figure 5A), comparable to the effect observed with DHMEQ. Moreover, mice in the NLRC5−/− + DHMEQ group exhibited the weakest macrophage phagocytosis, the lowest number of positive cells, and the least lipid and plaque deposition in the aortic tissue (Figure 5B), indicating a synergistic inhibitory effect of genetic NLRC5 silencing and pharmacologic NF-κB blockade. Histological analysis confirmed that this group showed the smallest atherosclerotic plaque area in the aorta (Figure 5C) and a significant increase in collagen fiber deposition (Figure 5D), indicating improved plaque stability. Together, these findings demonstrate that NLRC5 knockdown attenuates atherosclerotic progression in ACS by suppressing NF-κB-driven macrophage activity and inflammation.
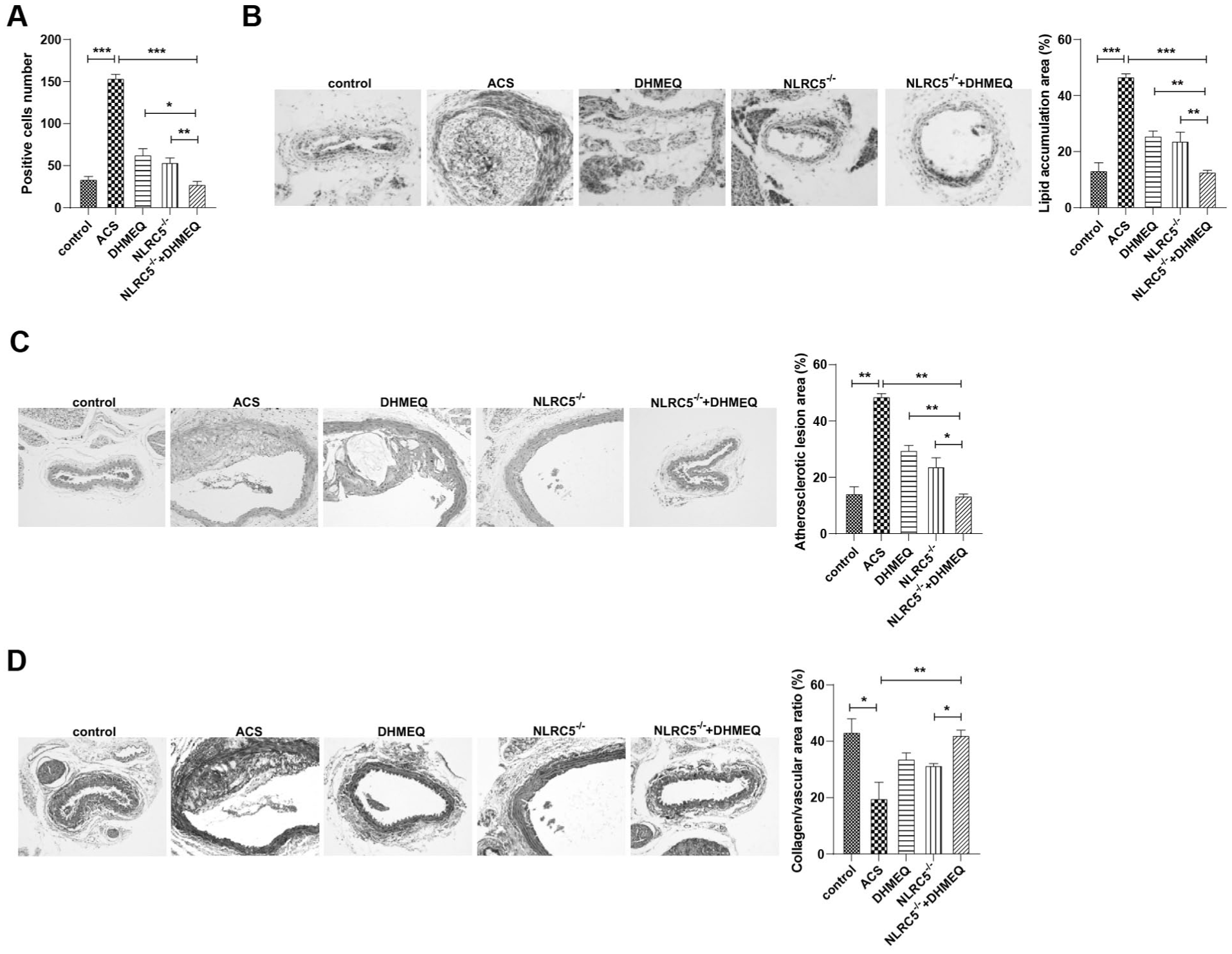
NLRC5 knockdown inhibits atherosclerotic plaque formation in ACS through modulation of the NF-κB cascade. (A) Ink phagocytosis assay to identify phagocytosis by macrophages. (B) Oil Red O staining to identify accumulation of lipids and plaques on the aorta. (C) HE staining to observe the cross-sectional area of arterial wall and atherosclerotic plaque area. (D) Masson staining to measure the aortic collagen area and calculate collagen/vascular area ratio. *P < .05, **P < .01, ***P < .001.
Discussion
AS, driven primarily by lipoprotein accumulation, leads to plaque formation at specific sites in the arterial wall, ultimately contributing to the pathogenesis of ACS. 30 A critical hallmark of plaque development is the transformation of circulating monocytes into lipid-laden macrophages, or foam cells, which contribute to lipid deposition and plaque instability.31,32 Lipid peroxidation and associated inflammatory responses further exacerbate plaque vulnerability by promoting apoptosis, inflammation, and neovascularization. 33 In this context, our study focused on the role of NLRC5 in foam cell formation and lipid accumulation, aiming to elucidate its contribution to atherosclerotic plaque progression in ACS.
We found that NLRC5 expression was significantly elevated in the peripheral blood of ACS patients, ~2-fold higher than in controls. This increase was accompanied by a notable reduction in methylation at the NLRC5 gene locus, suggesting epigenetic regulation of NLRC5 activity in ACS. Consistently, foam cells induced by acLDL also showed upregulated NLRC5 expression, further reinforcing its role in disease progression. As a member of the NLR family, NLRC5 functions as an innate immune sensor, and has been implicated in PANoptosis, a form of inflammatory cell death triggered by specific ligands. 34 NLRC5 upregulation and deubiquitination have been observed in murine atherosclerotic lesions, where its overexpression promotes endothelial-mesenchymal transition (EndMT) and accelerates plaque development. 35 Moreover, increased NLRC5 levels have been reported in circulating monocytes and cardiac macrophages in cardiovascular disease settings. 14 Beyond atherosclerosis, NLRC5 has also been linked to cardiac fibrosis, where its overexpression in fibroblasts promotes proliferation and myofibroblast transition, while NLRC5 silencing suppresses fibrotic remodeling. 36 In our study, NLRC5 knockdown in foam cells led to a reduction in inflammatory cytokine secretion and lipid accumulation, while in vivo, NLRC5−/− mice displayed lower lipid levels and attenuated inflammation in the context of ACS. These findings are in line with previous reports showing that NLRC5 silencing reduces inflammatory cytokines in various disease models, such as venous smooth muscle cells in varicose veins, 37 and in primary microglia and astrocytes exposed to neuroinflammatory stimuli. 38 Furthermore, NLRC5 overexpression has been associated with enhanced apoptosis in hypoxia/reperfusion-injured HL-1 cardiomyocytes and with the worsening of acute myocardial infarction. 39 Additionally, NLRC5 deletion protects against ethanol-induced hypertriglyceridemia and abnormal lipid metabolism, highlighting its broader role in lipid regulatory pathways. 40
NF-κB is a dimeric transcription factor composed of proteins from a 5-member family and plays a central role in regulating inflammation and immune responses by controlling the expression of chemokines, cytokines, transcription factors, and various effector proteins. 41 The pro-atherogenic nature of NF-κB has been well-established, as it is involved in key processes of atherogenesis, including foam cell formation, vascular inflammation, and plaque progression. 42 In this study, mechanistic investigations revealed that NLRC5 knockdown inhibited phosphorylation of p65 and inhibitor of κB (IκB) in foam cells, as evidenced by the reduced expression of p-p65 and p-IκB proteins. These findings suggest that NLRC5 facilitates the activation of the NF-κB cascade, consistent with prior reports.12,28 Furthermore, NLRC5 silencing was shown to suppress macrophage phagocytic activity, which was accompanied by a reduction in aortic plaque accumulation in ACS mice, suggesting that NLRC5 promotes plaque formation by regulating macrophage function through NF-κB signaling. This mechanism is further supported by previous findings. For example, reduction of methyltransferase-like 14 (Mettl14) levels has been reported to promote M2 macrophage polarization and inhibit foam cell formation via the NF-κB/IL-6 pathway. 43 Similarly, salvianolic acid B, under TNF-α stimulation, decreases reactive oxygen species production and prevents nuclear translocation of NF-κB, thereby attenuating atherosclerosis and inflammation via the NF-κB/NLRP3 pathway. 44 In another study, fullerene derivatives functioned as NF-κB inhibitors, effectively blocking foam cell formation and plaque accumulation. 45 Conversely, activation of NF-κB signaling has been shown to enhance macrophage infiltration, lipid accumulation, and foam cell development, ultimately reversing the anti-atherosclerotic effects of papain. 46
In conclusion, the present study demonstrates that NLRC5 contributes to atherosclerotic plaque formation in ACS by promoting macrophage-mediated inflammation and phagocytosis through activation of the NF-κB signaling pathway. NLRC5 knockdown not only reduces foam cell formation and inflammatory cytokine production but also inhibits the progression of lipid-rich plaques and enhances plaque stability. However, this study is subject to certain limitations. The downstream targets and broader signaling networks downstream of NF-κB were not fully explored. In addition, in vitro experiments to delineate the specific molecular interactions between NLRC5 and components of the NF-κB pathway remain lacking. Future studies should aim to identify the direct transcriptional targets of NLRC5-mediated NF-κB activation and assess the therapeutic potential of targeting this axis in both cellular and animal models.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Joint Program of Technology Research of Yunnan Provincial Science and Technology Department-Kunming Medical University, China (grant no 202001AY070001-060).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.