
Abstract
This narrative review aims to elucidate the effects of sleep deprivation (SD) on vascular endothelial cells and blood pressure, synthesize the mechanistic pathways involved, and propose potential intervention strategies, thereby positioning SD as a treatable target in hypertension management. A comprehensive literature search was conducted using databases including PubMed, Web of Science, Embase, and Medline for studies published up to October 2025. Evidence was systematically reviewed to examine SD-induced endothelial dysfunction and its role in blood pressure regulation. Both acute SD (including single-night total sleep < 7 hours, extended wakefulness up to ~36 hours, or ≤5 consecutive nights of restricted sleep) and chronic SD (persistent sleep restriction > 1 week) could induce measurable endothelial impairment and sustained blood pressure elevation. Key mechanistic pathways identified included: (1) vasomotor imbalance, (2) sympathetic nervous system hyperactivation with impaired baroreflex sensitivity, (3) systemic inflammation, (4) oxidative stress, (5) metabolic disturbances, and (6) circadian misalignment affecting vascular clock genes. SD is a significant, modifiable risk factor for endothelial dysfunction and hypertension. These findings highlight the therapeutic potential of endothelial-focused approaches and SD intervention in sleep-compromised populations. Future research should prioritize elucidating the underlying mechanisms and developing valid, cost-effective interventions.
Keywords
Introduction
For adults, the recommended sleep duration was 7 to 9 hours. 1 Sleep deprivation (SD) referred to a decrease in sleep or a severe lack of sleep resulting from various factors. 2 SD occurred when a person was not able to obtain sufficient sleep, defined as sleeping less than the duration recommended for health benefits. 3 SD was now a recognized health problem in this modern era, which was one of the major causes of visits to the sleep clinics as well. 4 Thirty-five percent of adults in the United States reported sleeping ≤7 hours during a typical 24-hour day. 5 It was estimated that 50 to 70 million Americans chronically suffered from a disorder of sleep and wakefulness, hindering daily functioning and adversely affecting health and longevity. 6
Short sleep duration was confirmed to be a risk factor for hypertension.7 -9 In clinical settings, ambulatory blood pressure monitoring (ABPM) parameters such as BP variability and nocturnal BP patterns provided important insights into cardiovascular risk and hypertension-related outcomes. 10 SD was capable of triggering different biological effects, such as neural autonomic control changes, 11 increased oxidative stress, 12 altered inflammatory 13 and coagulation responses, 14 and accelerated atherosclerosis, 15 as well as hypertension. 16 Meanwhile, SD was also reported to induce endothelial dysfunction. 17 For example, signal peptide-CUB-EGF domain-containing protein 1 (SCUBE-1), a biomarker associated with endothelial activation, was reported to be significantly elevated in hypertensive patients, particularly in those with hypertension-mediated organ damage, suggesting its potential role as an indicator of vascular injury. 18 However, the mechanisms by which SD affected endothelial cells (ECs) and led to hypertension were not clear, the present narrative review considered these points.
Literature Search
To support the synthesis of current evidence on the relationship between SD, endothelial dysfunction, and BP regulation, we conducted a structured literature search in PubMed, Web of Science, Embase, and MEDLINE, covering studies published up to October 2025. The search combined relevant MeSH terms and keywords, including “sleep deprivation,” “insufficient sleep,” “inadequate sleep,” “sleep loss,” “sleep insufficiency,” “sleep fragmentation,” “sleep debt,” “lack of sleep,” “not have enough sleep,” “endothelial*,” “vascular*,” “endothelial dysfunction,” “hypertension,” and “blood pressure.” Inclusion criteria were as follows: (1) peer-reviewed studies published in English, (2) studies investigating the mechanisms linking SD to endothelial function and/or BP regulation, and (3) both human and animal studies, as well as mechanistic reviews that provided insights into physiological pathways. Studies were screened at the title and abstract level, followed by full-text review for eligibility. Reference lists of selected articles were also manually searched to identify additional relevant publications not captured in the initial search. This review was conducted as a narrative review rather than a systematic review. Therefore, the selection of studies was primarily based on their relevance to the mechanisms linking SD, endothelial dysfunction, and BP regulation. Studies were selected to provide representative evidence across experimental, translational, and clinical research. Both human and animal studies were included to offer complementary mechanistic and clinical perspectives.
Mechanisms of Vasodilation-Contraction Imbalance Induced by Sleep Deprivation
Direct Effects on Contractile and Relaxation Imbalance of Endothelial Cells: Endothelin-1/Nitric Oxide (ET-1/NO)
The most intuitive manifestation of vascular endothelial dysfunction was the abnormal regulation of vascular tone. 19 The endothelium-derived constricting and relaxing factors jointly maintained the balance of vascular tone, thereby regulating BP. 20 The vasoconstrictor factors were ET-1, angiotensin II, thromboxane A2, thrombin, superoxide anion, and others. The vasodilative molecules included NO, prostaglandin I2, 21 endothelium-derived hyperpolarizing factor (Figure 1). 22
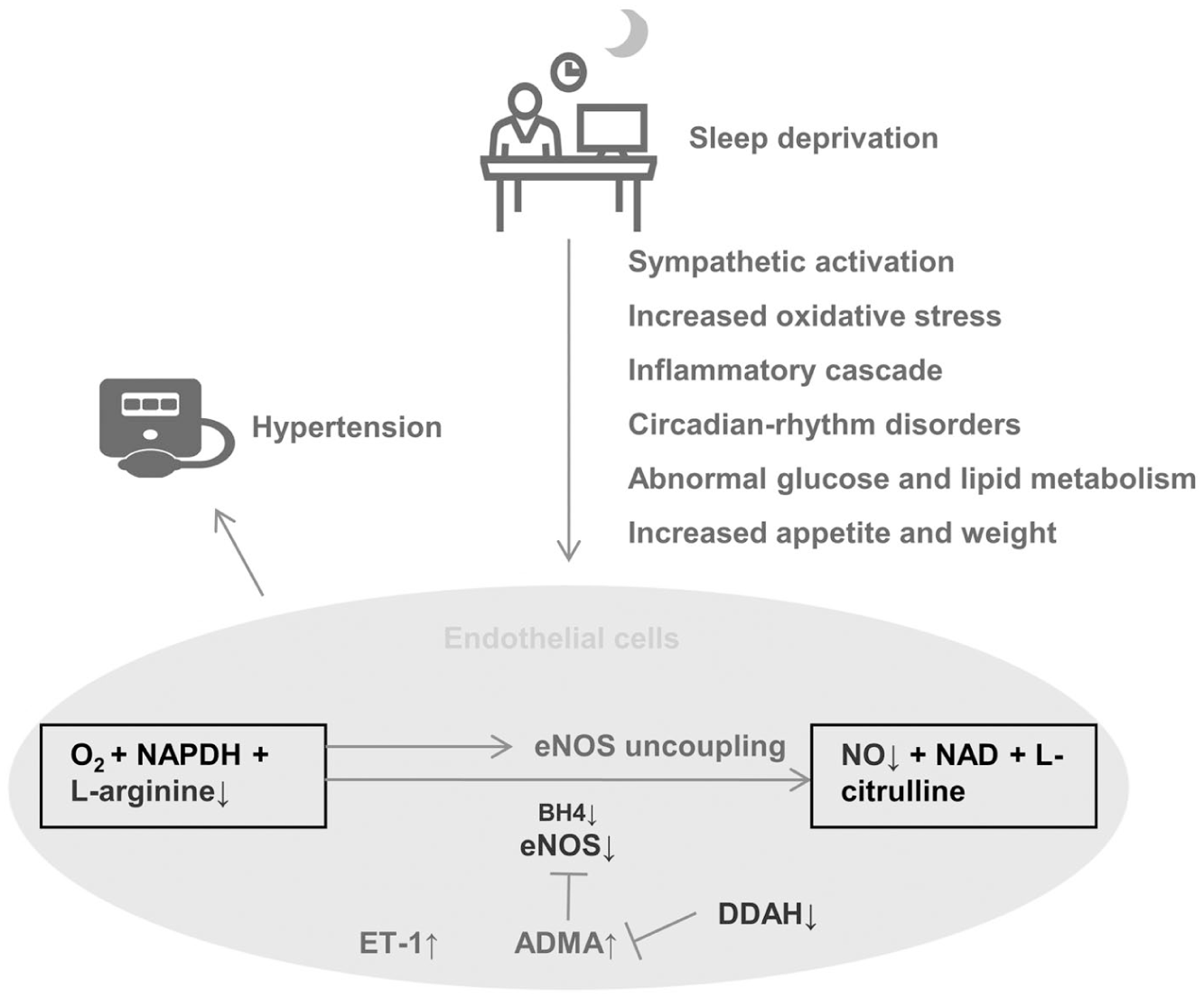
Possible mechanisms by which sleep deprivation (SD) impairs endothelial function and contributes to hypertension. SD induces sympathetic activation, oxidative stress, inflammation, circadian disruption, metabolic disturbances, and appetite changes, which collectively impair endothelial NO production by reducing eNOS expression (through decreased BH4, increased ADMA, and reduced DDAH), limiting substrate availability (L-arginine) and promoting eNOS uncoupling. Meanwhile, ET-1 expression is also increased. These alterations contribute to vascular dysfunction and hypertension.
ET-1 was an effective vasoconstrictive peptide, which played an important role in endothelial vasoconstriction. 23 After arterial perfusion with BQ-123 (a selective ET-A receptor antagonist) in the forearm blood flow of elder normotensive participants, the vasodilatory response of individuals who were short sleepers (6.1 ± 0.1 h/night) was 2.5 times (20% vs 8%, P < .05) higher than that of normal sleep group (7.6 ± 0.1 h/night), suggesting increased ET-1 activity during SD. 24 Using radioimmunoassay, 1 study found that plasma ET-1/2 levels in male Wistar rats increased significantly after 96-hour SD (6.58 fmol/mL) compared with controls (5.07 fmol/mL, P < .03) and the 24-hour SD group (~5.20 fmol/mL, P < .04), whereas 24-hour SD did not significantly alter ET-1/2 levels, suggesting that prolonged SD may be required to elevate plasma ET-1/2 levels. 25
There were 2 pathways 26 by which NO was produced in ECs, including endothelial NO synthase (eNOS) pathway and eNOS-independent pathway (nitrite could be either catalyzed to NO by oral facultative anaerobic bacteria and acidic gastric juice, or reduced by numerous proteins and enzymes in plasma such as hemeproteins, xanthine oxidoreductase, aldehyde oxidase and mitochondrial enzymes). 27 NO produced from L-arginine by the first pathway (eNOS pathway) was the primary source of blood NO. 26 The expression of eNOS in male Sprague–Dawley (6-8 weeks old) rats was reduced after suffering from 72-hour SD of rapid eye movement by using the inverted flowerpot technique—a well-established method in which animals are housed on small platforms surrounded by water, where the loss of muscle tone during rapid eye movement sleep causes them to fall and wake. 28 The decrease in NO inhibited sleep (non-rapid eye movement and rapid eye movement sleep) in rats.29,30 The vicious cycle of NO reduction and SD might contribute to the development of hypertension. In a study composed of hypertensive middle-aged and older adults, there was reduced acetylcholine-NO-mediated endothelium-dependent vasodilation and bradykinin-induced endothelial tissue plasminogen activator release in insufficient sleep group (5.8 ± 0.1 h/night) compared with the control group (7.6 ± 0.1 h/night). 31 In this study, the changes of endothelium-dependent vasodilation were mediated primarily by acetylcholine but not by bradykinin and nitroprusside, suggesting that sleep loss-induced hypertension may not involve impaired endothelium-dependent hyperpolarization as its underlying mechanism. 31 Bain et al. found that after injecting acetylcholine into the forearm artery of the elder normotensive male participants, the response to acetylcholine in individuals with short sleep (6.1 ± 0.2 h/night) was significantly diminished (4.6 ± 0.3 to 11.7 ± 1.0 vs 4.4 ± 0.3 to 14.5 ± 0.5 mL/100 mL tissue/min, ~20%, P < .05) compared with controls (7.7 ± 0.2 h/night). 32 Next, all the subjects were infused with the vasodilator inhibitor—N-monomethyl-L-arginine (a broad NO synthase inhibitor), but it only attenuated the response to acetylcholine of the control group (~40%). Therefore, the loss of endothelium-dependent vasodilation in SD group might be related to reduced NO bioavailability. 32 Asymmetric dimethylarginine (ADMA) is an endogenous inhibitor of NOS, which inhibits the synthesis of NO. 33 It was an independent predictor of cardiovascular mortality and morbidity. 34 Słomko et al 35 found that after experiencing 24-hour SD, 40 healthy male subjects (mean age: 31.2 ± 6.3 years; body mass index: 24.6 ± 2.6 kg/m2) exhibited a significant decrease in plasma L-arginine (68.8 ± 10.2 vs 78.2 ± 12.9, P = .001) compared with baseline along with an increase in ADMA (0.4 ± 0.1 vs 0.3 ± 0.1, P = .004), reflecting the disturbance of NO production of vascular ECs. Meanwhile, BP, especially systolic BP, remained elevated throughout the whole process. 35 These results provided potential mechanistic links between SD, impaired NO bioavailability, and elevated BP in humans. A study assessing sleep quality in patients with diabetes using the Pittsburgh Sleep Quality Index 36 classified those with an overall score > 5 as “poor sleepers.” The ratio of serum L-arginine/ADMA was decreased in poor sleepers compared with the good sleep group (35.1 [27.8-41.1] vs 42.0 [31.4-54.3], P < .01). The whole score was positively associated with the level of ADMA (r = .39, P < .01) and negatively with L-arginine/ADMA (r = −.28, P = .02). Notably, pure sleep duration was not significantly associated with either indicator (P > .05). 36 Hence, sleep quality, more than duration, appeared critical for L-arginine/ADMA/NO balance. Dimethylarginine dimethylaminohydrolase (DDAH) including DDAH1 and DDAH2, can catalyze the decomposition of ADMA, thereby regulating eNOS function and local NO concentration.37,38 It was beneficial to improve the progression of hypertension. 39 Meanwhile, increasing the cerebral level of DDAH not ADMA was proved to improve insomnia, which was regulated in part by oxidative stress.40,41 In basic research, Jiang et al 42 found that SD (5 days) could cause NO-induced endothelial dysfunction and an increase in BP (systolic BP: ↑16%; diastolic BP: ↑18%) in male middle-aged (24 weeks old) Sprague-Dawley rats by damaging the eNOS/NO/cyclic guanosine monophosphate pathway. SD was associated with decreased eNOS phosphorylation and reduced NO and cyclic guanosine monophosphate content in ECs of adult rats, but not in young (6-week-old) rats. 42 This finding was in line with the concept that vascular ECs are more vulnerable to damage with aging, 43 and that this damage might be amplified by SD.
Most mechanistic evidence was derived from animal models. Human studies remained largely observational, relying on functional or biomarker surrogates rather than direct molecular interrogation of the upstream regulatory mechanisms or downstream signaling cascades linking SD to endothelial dysfunction. Finally, whether SD alone could induce eNOS uncoupling, a mechanism well-documented in obstructive sleep apnea involving peroxynitrite overproduction and reduced tetrahydrobiopterin availability, remained an open question. 44
Sleep Deprivation Induces Sympathetic Activation
Sleep disorders and insufficiencies were often accompanied by heightened sympathetic activity, a known contributor to increased cardiovascular risk. 45 Activation of the hypothalamic–pituitary–adrenal axis and heightened sympathetic tone were also reported in the patients with insomnia and hypertension. 46 Heart rate variability could provide information about functioning of the autonomic nervous system and interaction of sympathetic and parasympathetic (vagal) pathways. 47 In a male cross-over study, sleep restriction (awake from 3:00 am to 7:00 am) not sleep fragmentization resulted in higher heart rate (P = .018) and lower pNN50 (percentage of RR intervals with >50 ms variation, a marker which reflected vagal activity, P = .012) during sleep stage N1 (non-rapid eye movement stage 1), as well as lower SDNN (standard deviation of RR intervals, a global marker for total heart rate variability, P = .009) during wakefulness compared with baseline. 48 Compared with non-shift workers, a significantly higher LF/HF (low frequency/high frequency, a marker which reflected sympathovagal balance), lower heart rate variability, and a trend for a lower flow-mediated dilatation (P = .08) were observed among shift workers required to stay awake for 30.5 hours, which reflected higher sympathetic and/or lower parasympathetic activities. 49 In a study of interventions for all, acute SD (36 hours) increased sympathetic activation, and decreased parasympathetic cardiovascular modulation and spontaneous baroreflex sensitivity in 18 healthy participants compared with baseline. 50 However, these human studies investigating autonomic responses to SD were conducted in relatively small experimental samples. In another cross-over study, 51 partial SD (3.5-5 hour) for 5 nights significantly increased sympathetic activity of 13 healthy Caucasian subjects compared with normal sleep condition (7-9.5 hour; serum norepinephrine [162 ± 58 vs 119 ± 46 ng/mL, P < .01]). Although the differences in BP rise (systolic BP: 112.9 ± 11.7 vs 108.8 ± 9.8 mmHg; diastolic BP: 65.3 ± 6.9 vs 63.6 ± 4.8 mmHg; P > .05) were not statistically significant, there was a decrease in endothelial-dependent venodilatation (maximal venodilatation [41 ± 20 vs 100% ± 22%, P < .001]) induced by acetylcholine. 51 Also, the mean dose of phenylephrine required to induce 20% venoconstriction was significantly reduced after SD (332 ± 107 vs 846 ± 189 ng/mL, P < .01). 51 Similarly, 52 the urinary excretion of norepinephrine was increased in 36 never-treated mild to moderate hypertensive patients suffering from 4-hour SD (awake from 11:00 pm to 3:00 am) compared with when they got enough sleep (2.12 ± .31 vs 1.57 ± .26 mmol/min, P < .05). In the next morning after SD, the BPs (systolic BP: 154.86 ± 5.91 vs 147.72 ± 6.84 mmHg, P < .001; diastolic BP: 96.29 ± 7.25 vs 92.28 ± 7.26 mmHg, P < .01) and heart rate (75.19 ± 6.33 vs 69.73 ± 5.26 beats/min, P < .05) remained elevated. 52 The divergence reflected differences between healthy individuals and patients with hypertension, suggesting that an intact endothelium in healthy subjects might initially buffer the hemodynamic consequences of sleep loss, whereas this compensatory capacity might be impaired once hypertension develops, rendering BP more susceptible to SD. Besides, the nocturnal BP phenotype of all hypertensive participants became non-dipping when experiencing SD, underscoring the heightened vulnerability of nocturnal BP regulation. 52 Interestingly, the BP (mean arterial pressure: 133.7 ± 2.8 vs 102.1 ± 3.8 mmHg, P < .05) and heart rate (500.9 ± 4.8 vs 416.5 ± 5.7 bpm, P < .05) were significantly increased in male Sprague–Dawley rats suffering from 21 days of chronic SD (awake for 20 h/day) compared with control rats. 53 The effects might be related to the upregulation of angiotensin II type 1 receptors in the nucleus tractus solitarii, contributing to impaired baroreflex sensitivity (pre-SD: −1.16 ± .13 bpm/mmHg vs post-SD: −.46 ± .12 bpm/mmHg, P < .05). Then, microinjection of losartan into the nucleus tractus solitarii reversed this damage of sleep-deprived rats (pre-intervention: −.46 ± 0.12 bpm/mmHg vs post-intervention: −1.01 ± .07 bpm/mmHg, P < .05). 53 Nevertheless, whether similar central mechanisms operate in humans exposed to chronic sleep restriction remained to be clarified.
Sleep Deprivation Leads to an Inflammatory Storm
The inflammation induced by sleep loss was attributed to glucocorticoids and catecholamines stimulating the activation of nuclear factor-kappa B to upregulate granulocytes and cytokines. 54 However, 1 study suggested that 1 week of sleep restriction (sleep from 2:00 to 6:00) directly impaired endothelial function through inflammatory pathways independent of sympathetic activation, and sympathetic activation under long-term SD only aggravated the inflammatory responses. 55 A recent study 13 suggested that SD increased brain-derived prostaglandin D2 efflux mediated by ATP-binding cassette transporter 4, which might cross the blood–brain barrier and trigger systemic inflammation. Interleukin-6 and interleukin-17A emerged as the most prominent pro-inflammatory cytokines induced by SD. 13 Interleukin-17 was mainly secreted by immune cells (T helper cells), 56 and its subtype interleukin-17A could reduce NO production by phosphorylating threonine 495 of eNOS in porcine aortic ECs. Systemic infusion of interleukin-17A was shown to increase BP in adult male C57BL/6 mice. 57 However, direct evidence linking interleukin-17-mediated pathways to SD-related hypertension in humans remains limited. Following 5 consecutive nights of partial SD (awake from 23:00 to 03:00), healthy male adults exhibited significantly elevated serum levels of interleukin-17A, interleukin-1β, interleukin-6, and C-reactive protein compared with baseline, along with increased heart rate which persisted until the following 2 nights of recovery sleep (P < .05). 58 Consistent with this, a recent prospective study in 14 healthy young men 59 found that 24-hour total SD significantly impaired endothelial function (flow-mediated dilatation: decreased from 6.7% ± 6.8% to 1.7% ± 3.3%, P = .009), accompanied by elevated salivary interleukin-1 (36 ± 21 to 47 ± 24 pg/mL, P = .004) and interleukin-6 (22 ± 7 to 36 ± 11 pg/mL, P = .0005). Notably, the decline in flow-mediated dilatation strongly correlated with the increase in interleukin-1 (r = −.813, P = .001) and interleukin-6 (r = −.735, P = .003), suggesting a direct link between acute inflammation and endothelial dysfunction. In contrast, the level of tumor necrosis factor alpha was reduced and BP remained unchanged throughout the process. 58 These findings suggested that inflammatory responses to SD may vary across cytokine pathways and experimental conditions. In addition, the BP elevation under SD could aggravate vascular shear stress, which increased the levels of activation markers in ECs (such as E-selectin and intercellular adhesion molecule-1), and activated ECs in turn induced more production of interleukin-6. 54 In healthy men suffering from 40-hour SD, the elevation of E-selectin level occurred nearly 7 hour before the onset of full sympathetic activation and BP rise, which reflected a greater sensitivity of endothelial dysfunction in the early stages of SD. 60 Several studies reported an increase in C-reactive protein after SD, both in the long-term sleep-deprived population and in normal sleepers with sudden sleep loss.61 -63 C-reactive protein was reported to interact with ECs, reduce the expression and bioactivity of eNOS and the production of NO, and increase the release of vasoconstrictors and adhesion molecules. 64 The role of C-reactive protein between SD and endothelial function was unknown. A meta-analysis showed that sleep disturbance and long sleep duration, but not short sleep duration, were associated with elevated systemic inflammatory markers. 65 However, most of the included studies were non-prospective and non-objective 65 studies, so the relationship between short sleep duration and systemic inflammation remains inconclusive. The inflammatory effect might be more obvious with the accumulation of SD and long-term circadian rhythm disorder, and more sensitive inflammatory markers were needed to verify this idea in the future. However, most mechanistic evidence originated from experimental and animal studies, and further clinical investigations were required to clarify the translational relevance of these findings.
Sleep Deprivation Leads to Oxidation/Antioxidation Imbalance
The body produces less reactive oxygen species and free radicals during sleep than during wakefulness. 66 They could not only directly inhibit the bioactivity of NO, but also stimulate the phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B)-mitogen-activated protein kinase (MAPK) pathway related to redox transcription factors, which might lead to overexpression of redox genes and subsequent inhibition of eNOS mRNA expression and its bioactivity. 67 Antioxidant molecules and enzymes such as reduced glutathione, catalase, glutathione peroxidase, superoxide dismutase, seemed active in sleep, helping to reduce oxidative stress, 68 but this increase remains controversial. 69 Compared with the control group, there was a significant decrease in glutathione levels but an increase in thiobarbituric acid reactive substance (an index of lipid oxidation indicating the levels of malondialdehyde in the tissues) levels and catalase activity in the plasma of rapid eye movement sleep-deprived (96 hours) juvenile but not adult male Wistar rats. 70 Similarly, Nawi et al 71 found that the activity of superoxide dismutase was significantly reduced but malondialdehyde concentrations were increased in male Sprague–Dawley rats of rapid eye movement sleep-deprived (72 hours) compared with control rats. The resulting accumulation of superoxide anion might induce lipid peroxidation, which might contribute to aortic endothelial dysfunction and increased systolic BP. Furthermore, SD was associated with accumulation of reactive oxygen species and consequent oxidative stress, specifically in the gut. 12 The accumulation of reactive oxygen species might lead to upregulation of nuclear factor-kappa B in the gut and potential damage to intestinal barrier function (the levels of 2 critical tight junction proteins in the ileum of SD mice, zonula occludens-1 (ZO-1) and occludin were decreased), probably in turn aggravating systemic inflammation. 72 However, there was no reliable evidence that the changes of intestinal flora under SD-induced BP elevation.73,74 Only one intestinal microbial function module—2-aminoethylphosphonate transport system was observed to be simultaneously deficient in both normotensive and hypertensive subjects who slept short (≤6 hours) duration/day. 74 Sleep-related deoxycholic acid was also found to be upregulated in intestinal fecal metabolites in older people with short sleep duration, 75 and the deoxycholic acid was proved to attenuate phenylephrine-induced vasoconstriction in arterioles of mice and BP rise in male C57BL/6 specific pathogen-free (SPF) mice. 76 Nevertheless, current evidence in humans remains limited and further clinical validation is needed.
Hyperlipidemia and Hyperglycemia
SD was closely associated with the prevalence of metabolic syndrome.77 -79 Metabolic disturbances induced by SD might further exacerbate endothelial dysfunction and contribute to hypertension development. SD not only was associated with hypertension, but also with hyperglycemia (insulin resistance) and dyslipidemia.80 -86 However, the relationship between SD and dyslipidemia appeared to be complex and might be influenced by oxidative stress and inflammatory pathways.85,87 The coexistence of hyperglycemia and hyperlipidemia was characterized by activation of the ET-1 system and reduced eNOS and nuclear factor erythroid 2-related factor 2 (Nrf2, an antioxidant factor) activity, which might contribute to endothelial dysfunction. 88 Kong et al 89 reported that high-fat/high-glucose conditions could exacerbate the endothelial insulin resistance and dysfunction, accompanied by decreased NO levels, elevated ET-1 levels, weakened PI3K/Akt/eNOS signaling, and impaired endothelium-dependent vasodilation function. 89 Among hypertensive patients with metabolic syndrome, there was likely a high prevalence of target-organ damage. 90 Therefore, improving sleep duration and quality represented an important strategy for preventing hypertension and metabolic disorders.
Obesity, Leptin, and Ghrelin
Alterations in appetite-regulating hormones, particularly leptin and ghrelin, were implicated in obesity and metabolic dysregulation, which were closely associated with hypertension.91,92 In the Wisconsin sleep cohort study, short sleep duration was associated with reduced leptin, elevated ghrelin, and increased body mass index. 93 Ghrelin was found to activate AMPK signaling in angiotensin II-induced hypertensive male C57BL/6 mice, leading to inhibition of oxidative stress, increased NO production, improved endothelial function, and reduced BP. 94 In addition, although chronic not acute hyperleptinemia and leptin resistance were associated with hypertension and endothelial dysfunction, these effects were in part related to obesity and gender.95 -97 These hormonal disturbances caused by SD might lead to increased caloric intake, particularly from energy-dense foods rich in fat and carbohydrates, 98 thereby promoting weight gain and metabolic dysregulation. Such a high-calorie diet was shown to promote NO inactivation, ROS production and BP elevation in female Fischer rats. 99 Furthermore, it was well established that endothelial dysfunction in obesity-associated hypertension was related to NO bioavailability, oxidative stress, sympathetic activation, renin-angiotensin system, insulin resistance, as well as leptin resistance. 100
Circadian Rhythm Gene Variations in Sleep Deprivation-Related Vascular Dysfunction
The circadian rhythm of mammals consisted of a central clock and peripheral clocks. The central clock is located in the suprachiasmatic nucleus, and the peripheral clocks exist in almost every tissue. 101 Clock-controlled genes acting on the cardiovascular system could regulate various physiological functions of the circulation, such as endothelial function, BP, heart rate, acute myocardial infarction and the onset of arrhythmia. 102 Circadian rhythms were controlled by clock genes, which formed an oscillatory mechanism that was based on a self-sustaining transcription-translation feedback loop consisting of positive and negative feedbacks (Figure 2). 103
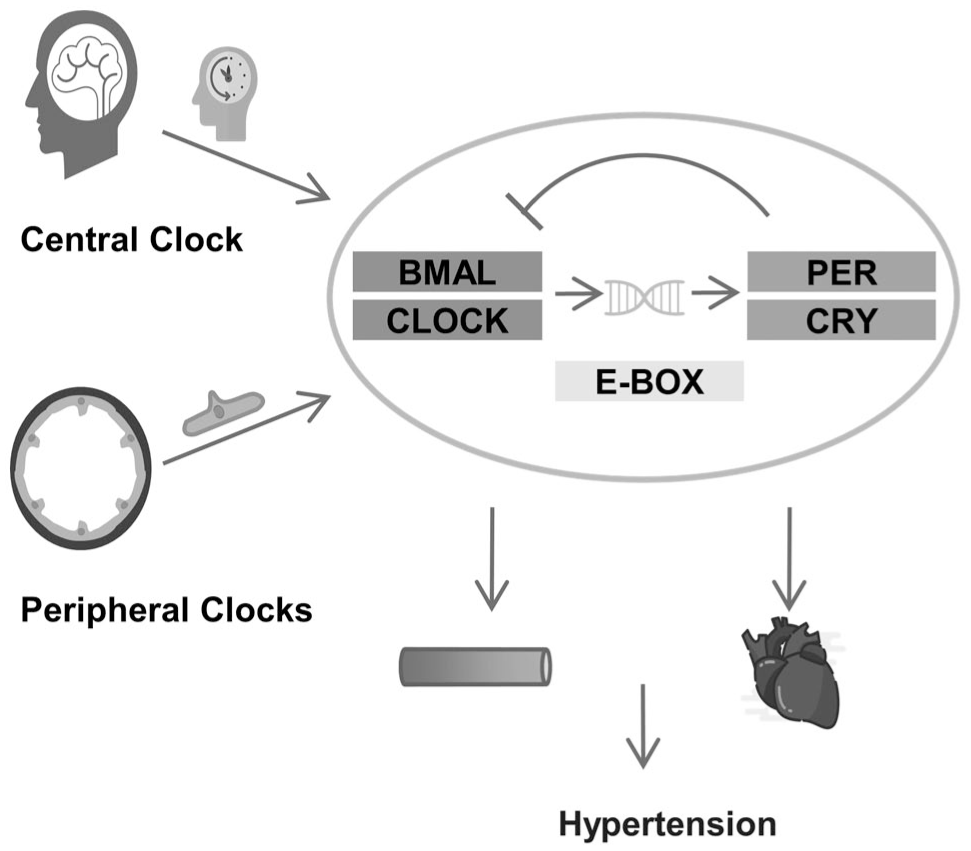
The association of circadian rhythm genes with endothelial cells (ECs) and blood pressure (BP). The regulation of circadian rhythms involves both the central circadian clock and peripheral endothelial clocks. At the molecular level, a BMAL/CLOCK–PER/CRY negative feedback loop (mediated by E-box) drives 24-hour oscillations in gene expression. Dysregulation of this loop may disrupt endothelial homeostasis, thereby promoting vascular dysfunction and hypertension.
The reduced time spent sleeping disrupted the circadian time structure. 104 Chronic SD (2 months) resulted in abnormal expression of brain and muscle ARNT-like 1 (BMAL1), circadian locomotor output cycles protein kaput (CLOCK), and cryptochrome 1 (CRY1) in the circadian rhythm-related nuclei of experimental mice. 105 In aortic ring tissue bath studies, the acetylcholine induced 73% ± 1.9% relaxation in wild-type mice but this response was severely blunted to 26.9% ± 1.7% in BMAL1-knockout mice (P < .05). In this process, eNOS was observed to be uncoupled in aorta characterized by reduced levels of tetrahydrobiopterin, elevated dihydrobiopterin levels, reduced tetrahydrobiopterin:dihydrobiopterin ratio, and production of superoxide in place of NO. 106 Naive aortas from BMAL1-knockout and CLOCK mutant mice both exhibited an impaired endothelium-dependent vasodilator response to acetylcholine, but the vasodilation in the latter was also related to the conditions of light-dark and dark-dark. 107 Moreover, the downregulation of endothelium-derived vasodilation in BMAL1-knockout mice was associated with attenuated phosphoinositide dependent kinase/Akt/eNOS signaling. 107 Compared with control group, SD (awake for 20 h/day for 7 days) increased the expression of pro-inflammatory cytokines and adhesion molecules, activated cyclic adenosine monophosphate/protein kinase A/nuclear factor-kappa B and the binding of monocytes to ECs, while reducing the expression of CRY1 in male C57BL/6 mice ECs. 108 However, overexpression of CRY1 in ECs could reverse this vascular inflammation. 108 Okamura et al 109 reported that mice lacking both CRY1 and CRY2 exhibited salt-sensitive hypertension with hyperaldosteronism. In addition, mutation in the period 2 (PER2) gene in mice was associated with aortic endothelial dysfunction involving decreased production of acetylcholine-induced NO and vasodilatory prostaglandins I2, and increased release of cyclooxygenase-1-derived vasoconstrictors. 110 Interestingly, BP was reduced in the mutant mice compared with the control group, suggesting that the parasympathetic nerve or acetylcholine-mediated signaling in the heart of the PER2 gene mutant mice remained intact. 110 In PER2 mutant mice, there was significantly increased Akt signaling which was associated with increased oxidative stress and reduced eNOS activation, which induced vascular senescence and decrease in NO bioavailability. 111 It was also reported that global PER2 mutant and BMAL1 mutant mice showed lower BP but the endothelial-specific deletion of BMAL1 did not affect the variation in BP. 112 In short, there was no clear evidence on the effect of these circadian genes, which sometimes even manifested a hypotensive phenotype after being completely removed.113,114 These observations highlighted the intricate role of circadian clock genes in coordinating vascular signaling and BP regulation, with systemic and endothelial circadian mechanisms potentially playing distinct roles.
Interventions to Improve Endothelial Function During Sleep Deprivation
Exercise Training
Continued physical exercise was shown to alleviate impairment of reactive hyperemia in patients with essential hypertension through increased release of NO,115 -117 although increased physical activity might be offset by the negative consequences of chronically disturbed sleep. 118 A meta-analysis revealed that moderate continuous aerobic training (50% PO2max) for 30 to 40 min/session at least 3 times/week, was effective to improve endothelium-dependent vasodilation in hypertensive individuals. 119 In healthy young (17-30 years old) adults, completing the aerobic exercise (a single 50-minutes bout of cycling exercise) before acute restricted sleep (3.5 hours) could attenuate the increased 24 hours ambulatory systolic BP but not restore it to baseline, and this BP-lowering effect only occurred in participants identifying as late chronotypes (P = .045). 120 This finding suggested that exercise may partially offset the vascular effects of acute sleep restriction, although it may not fully normalize BP. It was reported that compared with before training, 7 weeks of a moderate to high-intensity interval training 121 prevented the decrease of acetylcholine-induced vasodilation in healthy young (27.3 ± 5.4 years old) males experiencing 40-hour SD. In the absence of significant changes in BP, pro-inflammatory cytokines such as interleukin-6 and tumor necrosis factor-α, vascular endothelial markers (monocyte chemoattractant protein-1 and E-selectin), and pulse wave velocity seemed to have changed earlier under SD. 121 Fortunately, exercise training improved the endothelial dysfunction of the participants. 121 In addition, compared with the normal sleep and exercise group and the pure normal sleep group, a session of high-intensity interval exercise to expend 500 kcals of energy, 122 even after acute SD (3-3.5 hours), could still maintain the brachial artery flow-mediated dilatation of the healthy male (31 ± 5 years old) participants. However, moderate-to-vigorous intensity exercise after SD might be detrimental to cardiovascular health. In contrast, the habit of regular exercise might mitigate the adverse effects of SD (Figure 3). 123
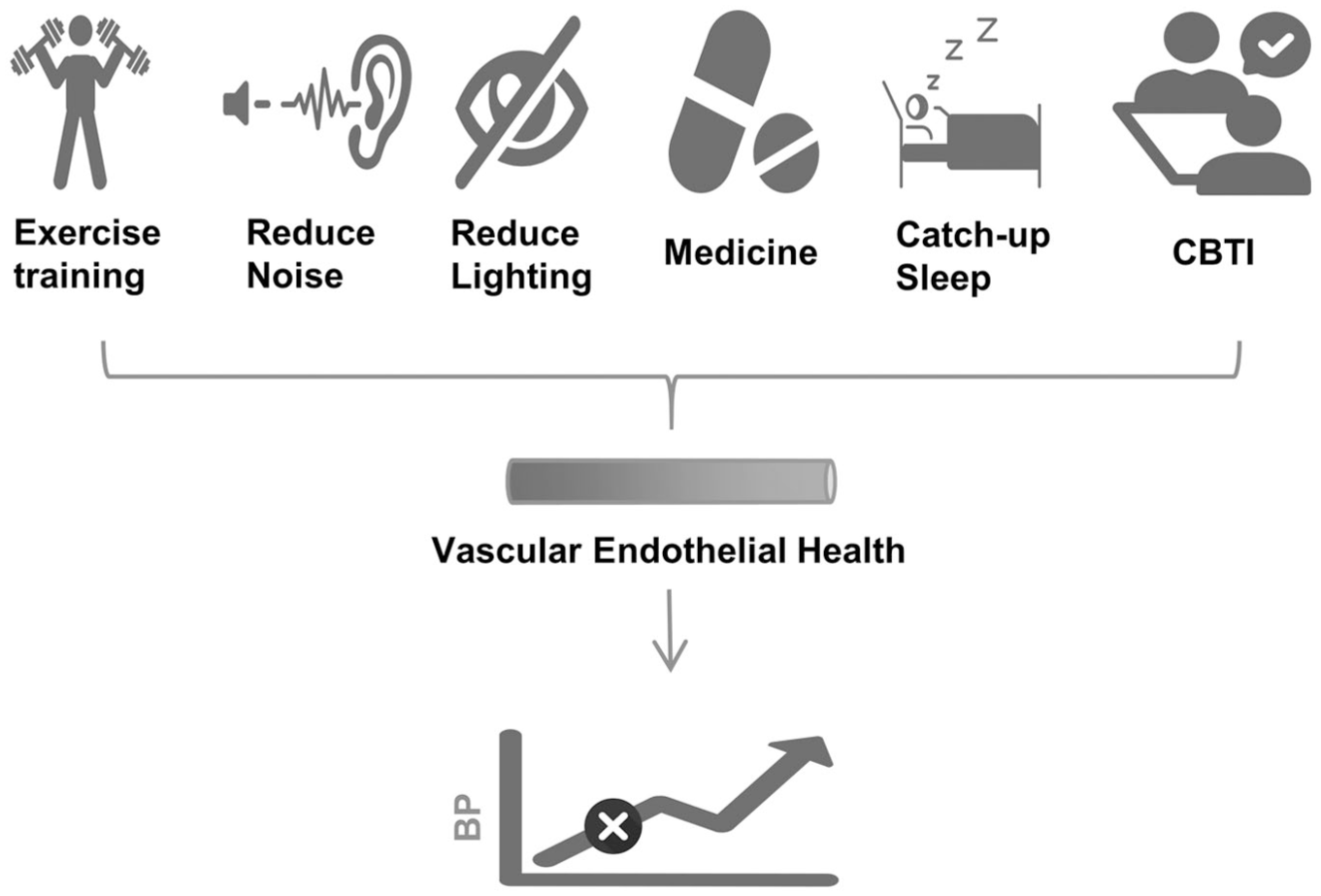
The possible options for addressing endothelial dysfunction caused by sleep deprivation (SD).
Noise Reduction
Environmental noise was an important factor contributing to sleep disturbance and SD. Exposure to noise levels of >75 dB over 1 year could result in poor sleep efficiency and short sleep duration. 124 Nocturnal noise affected not only autonomic regulation (increased heart rate and BP mediated by sympathetic activation and/or parasympathetic withdrawal), but also vascular function through the induction of endothelial dysfunction. 125 When exposed to 3 nighttime noise scenarios—background noise in home, 30 or 60 train noise events, with average sound pressure levels of 33, 52, and 54 dB, respectively, the flow-mediated dilatation of the brachial artery was gradually decreased in 70 healthy volunteers (11.23% ± 4.68%, 8.71% ± 3.83%, and 8.47% ± 3.73%; P < .001). 126 Mean sound level of 72 dB applied by loudspeakers for 4 days caused an increase in systolic and mean BP in mice, as well as endothelial dysfunction. 127 Noise exposure was associated with eNOS uncoupling in ECs, characterized by increased eNOS expression, elevated vascular tyrosine nitration in the endothelial layer, and eNOS S-glutathionylation, ultimately leading to reduced NO levels. 127 Interestingly, Vitamin C was reported to partially reverse the nocturnal noise-induced reduction in flow-mediated dilatation due to excessive oxidative stress. 128
Other Protective Measures
Several nutritional, behavioral, and therapeutic strategies were developed to mitigate endothelial dysfunction associated with SD. The supplementation with L-arginine was shown to protect against SD-induced hypertension and endothelial dysfunction (attenuated NO-mediated vasodilation), and upregulated the eNOS/NO/cyclic guanosine monophosphate pathway in middle-aged Sprague–Dawley rats. 42 It was also reported that flavonoids might improve endothelial function by increasing NO bioavailability, leading to relaxation of the endothelial smooth muscle, thus controlling vascular tone and BP. 129 In healthy individuals (25.31 ± 3.60 years old) experiencing 1 night of SD, the flavanol-rich group had higher level of flow-mediated dilatation (7.04 ± .62 vs 5.00% ± .49%, P < .001) and lower BPs compared with flavanol-poor group (systolic BP: 116.9 ± 1.6 vs 120.8 ± 1.9 mmHg, P < .001; diastolic BP: 70.5 ± 1.2 vs 72.3 ± 1.2 mmHg, P = .01). 129 In addition, the melatonin might increase endothelium-dependent vasodilation by inhibiting calcium channels and stimulating eNOS pathways. 130 However, exposure to nocturnal light could lead to inhibition of melatonin secretion.131 -133 A cross-sectional study even found that nocturnal light at home was significantly associated with increased nighttime BP in elderly individuals independently of overnight urinary melatonin excretion. 134 Wearing blue light glasses could block nocturnal light and maintain secretion of melatonin. 135 However, the benefits of melatonin on BP were debatable. 136 Two cross-sectional studies also showed that catch-up sleep (sleep 1-2 hours more on weekends than on weekdays) could reduce the risk of hypertension.137,138 The BP-elevating effect of short sleep duration could be reversible by 6 weeks of increased habitual sleep duration (Figure 3). 139 Slow-breathing training that stimulated parasympathetic activation was also reported to improve endothelial function and BP, particularly in those without SD. 140 Cognitive behavioral therapy demonstrated significant efficacy in the management of primary insomnia. 141 Whether cognitive behavioral therapy is beneficial in improving markers of cardiovascular diseases such as endothelial function in people with insomnia and hypertension, was currently being investigated in clinical trials. 142 A meta-analysis showed that cognitive behavioral therapy could improve systolic BP (−8.67 mmHg, P < .001), diastolic BP (−5.82 mmHg, P < .001), sleep quality and negative mood in patients with hypertension. 143 SD was regarded as a stressor, 144 and cognitive behavioral therapy could also improve the anxiety and depression symptoms associated with insomnia, 145 which had multiple therapeutic effects. In addition, the most commonly used sedative-hypnotics in insomnia had both endothelium-dependent and endothelium-independent vasodilatory effects, 146 but their impact on endothelial function and BP in sleep‑deprived individuals remained unclear. A recent meta‑analysis of randomized controlled trials indicated that hypnotics did not significantly affect nocturnal or 24-hour BP. 147 Notably, prospective data revealed that the use of benzodiazepines and nonbenzodiazepines was associated with an increased risk of future cardiovascular events.148,149 The 2017 American Academy of Sleep Medicine clinical practice guidelines for insomnia 150 recommended cognitive‑behavioral therapy for insomnia as first-line treatment, with pharmacologic hypnotics reserved for patients who did not benefit from non-pharmacologic measures, used for limited durations and monitored closely, particularly in those at risk for cardiovascular events. Therefore, although sedative‑hypnotics improved sleep symptoms in selected patients, their routine use to mitigate SD-related endothelial dysfunction or hypertension was not supported by current evidence and should be undertaken with caution.
Conclusions
Based on classical hypertension mechanisms, this review systematically summarized the functional alterations of vascular ECs under SD and proposed potential therapeutic interventions. As the primary vascular barrier, ECs play an important role in BP regulation. Despite their crucial functions, fundamental research investigating the impact of SD and circadian rhythm disruption on vascular ECs and hypertension remains limited. It is worth noting that the current clinical studies on sleep and hypertension also have some shortcomings. 151 Future research should further clarify which types of SD (reduced sleep duration, sleep fragmentation, or circadian misalignment) exert the greatest impact on endothelial function and BP regulation. In addition, integrating endothelial biomarkers and ABPM parameters into clinical studies may help better characterize SD-related vascular alterations and identify populations that are particularly vulnerable to SD-induced cardiovascular risks. Moreover, developing effective strategies to mitigate and potentially reverse SD-induced vascular damage represents an important direction for future research.
Footnotes
Ethical Considerations
As a narrative review that synthesizes data and conclusions exclusively from publicly available papers, ethical approval is not applicable.
Author Contributions
Yu Yan: Conceptualization, Methodology, Investigation, Writing – original draft, Writing – review & editing. Dan Yang: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. Shuangliang Ma: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. All authors contributed to: (1) substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data, (2) drafting the article or revising it critically for important intellectual content, and (3) final approval of the version to be published.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Declaration of Generative Artificial Intelligence (AI) and AI-Assisted Technologies
No AI techniques were used for writing.