
Abstract
Objective:
To identify and characterize hearing loss (HL) in children with septo-optic dysplasia (SOD).
Methods:
Otologic and audiometric data for patients less than 18 years of age identified as having SOD who were seen in the Children’s Healthcare of Atlanta-Scottish Rite Hospital clinic between 2013 and 2017 were collected and reviewed through a HIPAA-compliant medical record search. Relevant literature was also reviewed with the assistance of Medline.
Results:
Sixty-four patients with SOD were identified, and 7 of those patients (10.9%) were diagnosed with hearing loss. Type of hearing loss was sensorineural (SNHL) in 5 patients (63%), mixed (MHL) in 1(14%), and conductive (CHL) in 1(14%). Bilateral loss presented in 60% (3/5) of SNHL patients, while the rest demonstrated unilateral loss. Unilateral findings included cochlear nerve deficiency (1) and atresia/microtia (1). Tympanostomy tubes were required in 57% (4/7) of SOD children with hearing loss. Amplification was successfully implemented in 86% (6/7).
Conclusions:
Hearing loss was found in nearly 11% of SOD children, and SNHL was identified as (63%) the predominant form of loss. To our knowledge, this is the first retrospective review of hearing loss in a pediatric SOD cohort and the first to report of cochlear nerve deficiency and atresia/microtia in this population. Based on these findings, early identification of hearing loss with imaging when appropriate and treatment of otitis in this population is recommended.
Introduction
Septo-optic dysplasia (SOD), also known as de Morsier syndrome, is a congenital developmental anomaly highlighted by septum pellucidum agenesis, optic nerve hypoplasia, and hypopituitarianism. 1 First described by Reeves 2 in 1941, this condition was later termed septo-optic dysplasia by de Morsier 3 in 1956. SOD exhibits an incidence of 1 in every 10 000 live births and presents equally in males and females. 4 While HESX1, SOX 2, and SOX3 mutations have been associated with SOD, none have been demonstrated as pathogenically linked to SOD. 5 The phenotypic presentation of SOD varies widely, but the 3 characteristic clinical manifestations are (1) anterior midline brain abnormalities (septum pellucidum, corpus callosum, anterior commissure), (2) optic nerve hypoplasia, and (3) hypothalamic-pituitary insufficiency.1,3 Other SOD-associated features include seizures, cerebral palsy, sleep disturbance, precocious puberty, obesity, anosmia, cardiac anomalies, and hearing loss.1,5
Permanent childhood hearing loss is among the most common congenital disorders, occurring in 1 to 3 per thousand births and 2 to 4 per 100 neonatal intensive care unit (NICU) graduates. 6 The advent of universal newborn hearing screening has revolutionized the early identification of affected neonates. While hearing loss has been associated with other phenotypically similar but genetically distinct congenital neurological disorders like optic nerve hypoplasia (ONH) and congenital pituitary hormone deficiency (CPHD), no prior studies have specifically examined hearing loss in the SOD population.7-9 Early identification of hearing loss in neonates with visual loss may also lead to the earlier identification of congenital neurological disorders like SOD, ONH, or CPHD. Although hearing loss is sometimes referred to as a potential associated feature of SOD, to our knowledge, no studies have examined the prevalence and characteristics of hearing loss in children with SOD. 1
Methods
Medical records of patients diagnosed with SOD seen in our multidisciplinary pediatric otolaryngology clinic at Children’s Healthcare of Atlanta-Scottish Rite Hospital between January 1, 2013, and December 31, 2017, were retrospectively reviewed. Charts of patients with SOD were identified through a HIPAA-compliant database and evaluated. Parameters reviewed included patient demographics, clinical characteristics, audiologic evaluations, and related neuroimaging studies. Data collected from audiometric evaluations consisted of pure tone audiometry, speech awareness thresholds, and tympanometry. Masked results, if present, were utilized when appropriate to ensure accurate threshold identification. If children were unable to complete regular audiometric testing and were evaluated by automated brainstem response testing (ABR) instead, then the resulting data from ABR were collected. All patients whose records were reviewed had previously received neuroimaging via magnetic resonance imaging (MRI) as part of their SOD diagnosis, and dedicated temporal bone imaging via MRI and/or computed tomography (CT) was obtained in children with hearing loss. These images were used to examine temporal bone anatomy for anomalies and/or malformations. Relevant literature was examined with the assistance of Medline. However, given the limited body of available research and the descriptive nature of this study, on the manifestations of hearing loss in pediatric patients, a thorough systematic review was not feasible. Institutional Review Board approval was obtained from Children’s Healthcare of Atlanta Institutional Review Board for this study.
Results
Sixty-four patients with clinically diagnosed SOD were identified through a HIPAA-compliant computer database search; 7 of these patients (10.9%) were also diagnosed with hearing loss based on Universal Hearing Screenings and follow-up audiometric confirmation. Of these 7 patients, 71% were male, with an age range between 13 months and 12 years at the time of database identification. All 7 SOD patients with HL manifested SOD stigmata, including agenesis of the corpus callosum/septum pellucidum, optic nerve hypoplasia, and pituitary insufficiency. All patients were developmentally delayed, ranging from mild to severe, and were diagnosed with SOD during the first few months of life through imaging as part of their neuroendocrine evaluations. One patient in this group died from unrelated causes at 20 months of age.
Pure sensorineural hearing loss (SNHL) was identified in 7.7% (5/64) of our SOD cohort and in 71% (5/7) of those with identified hearing loss (Table 1). Otoacoustic emissions (OAE) were absent in all ears with hearing loss and were present in ears without loss. Sixty percent (3/5) of the patients with SNHL demonstrated bilateral loss. Patients with unilateral loss were equally split between right and left ear. Unilateral SNHL was identified in 2 children (3.1% of the total SOD cohort), while unilateral mixed hearing loss (MHL) and conductive hearing loss (CHL) were each identified in 1 patient (1.6% of the total cohort). Of the 2 SOD patients with a CHL component, 1 was due to atresia, while the other was presumed to be ossicular in origin. No subjects demonstrated progressive loss. Audiograms demonstrated a flat configuration across all tested frequencies.
Patient Characteristics.
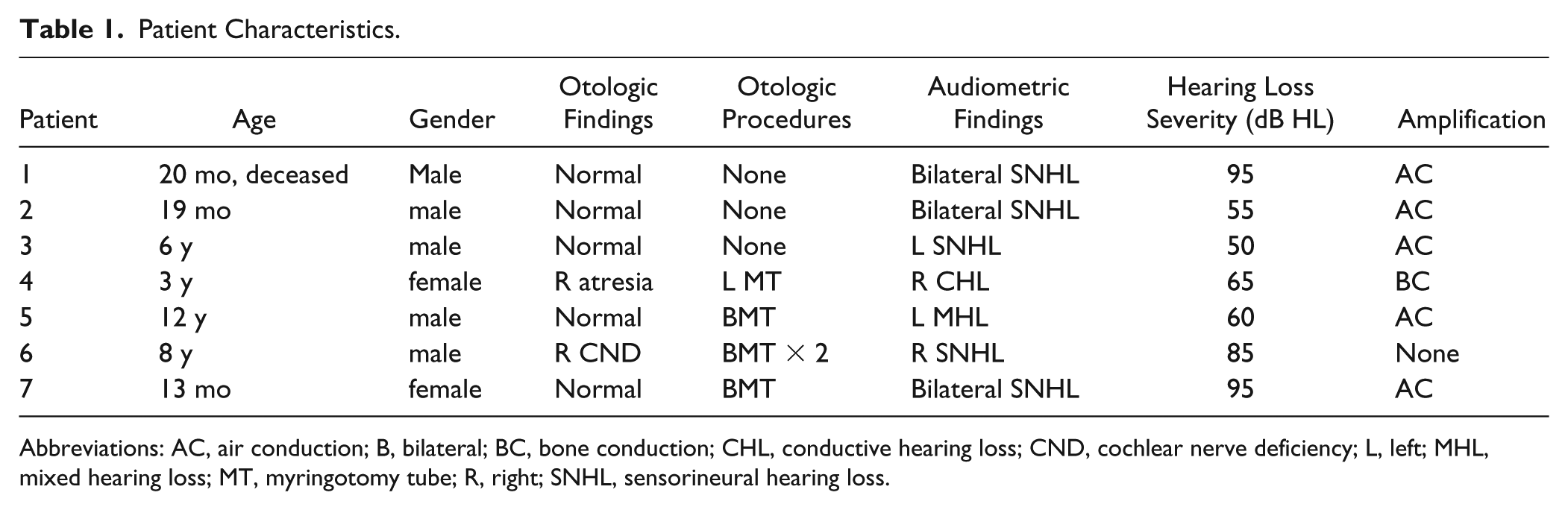
Abbreviations: AC, air conduction; B, bilateral; BC, bone conduction; CHL, conductive hearing loss; CND, cochlear nerve deficiency; L, left; MHL, mixed hearing loss; MT, myringotomy tube; R, right; SNHL, sensorineural hearing loss.
Neuroimaging with MRI was performed in all SOD patients. Temporal bone abnormalities included cochlear nerve hypoplasia (1/7) and aural atresia (1/7). No isolated cochleovestibular malformations were identified in SNHL SOD children to account for their loss. Collectively, significant temporal bone malformations were noted in 3.1% (2/64) of SOD patients in this cohort and 29% (2/7) of SOD patients with diagnosed hearing loss.
Level of hearing was defined as normal (0-19 dB HL), slight loss (20-29 dB HL), mild loss (30-39 dB HL), moderate loss (40-64 dB HL), severe loss (65-90 dB HL), and profound loss (over 90 dB HL). During the time interval of this study, hearing losses did not demonstrate fluctuations or progression. As demonstrated in Table 1, analysis of the most recent audiometric data revealed moderate (3), severe (2), and profound (2) hearing losses in this patient group. Audiometric thresholds were determined by averaging 4 frequency pure tones at 500 Hz, 1 kHz, 2 kHz, and 4 kHz. All audiometric thresholds were obtained with either clear middle ear spaces or patent tympanostomy tubes, reflecting persistent hearing losses despite optimized middle ear status.
Amplification was utilized in 86% (6/7) of SOD patients with HL. The predominance of air conduction amplification (5/6) reflects normal outer ear and canal anatomy of this population. Four children with SOD had 4 different amplification experiences. The first child with unilateral SNHL did not tolerate attempts with amplification. The second child was recommended for bone conduction amplification because of atresia, which was well tolerated. The third child met criteria for cochlear implantation, but the family deferred due to the severity of associated comorbidities and severe developmental delay. The fourth child, with unilateral cochlear nerve deficiency, was not offered ABI due to serviceable hearing with amplification in the contralateral ear.
Fifty-seven percent (4/7) of patients with SOD and hearing loss received tympanostomy tubes for chronic otitis media, and 14% (1/7) received more multiple sets. Only 57% (8/13) demonstrated normal tympanometry (Table 2) at the time of their last evaluation, suggesting a need for ongoing surveillance. One ear was not tested due to atresia.
Tympanometric Data.

Abbreviations: A, normal; B, middle ear effusion and/or functional myringotomy tube; C, negative middle ear pressure/ tympanic retraction; CNT, cannot test.
Discussion
Septo-optic dysplasia is a rare congenital disorder characterized by malformations of midline brain structures, including the hypoplasia or dysplasia of the corpus callosum/septum pellucidum, hypothalamic/pituitary dysfunction, and hypoplasia of the optic nerves (Figure 1). Significant variability in presentation exists, with only 30% of affected patients demonstrating the complete SOD triad and 40% showing normal endocrine function despite presence of imaging abnormalities. 10
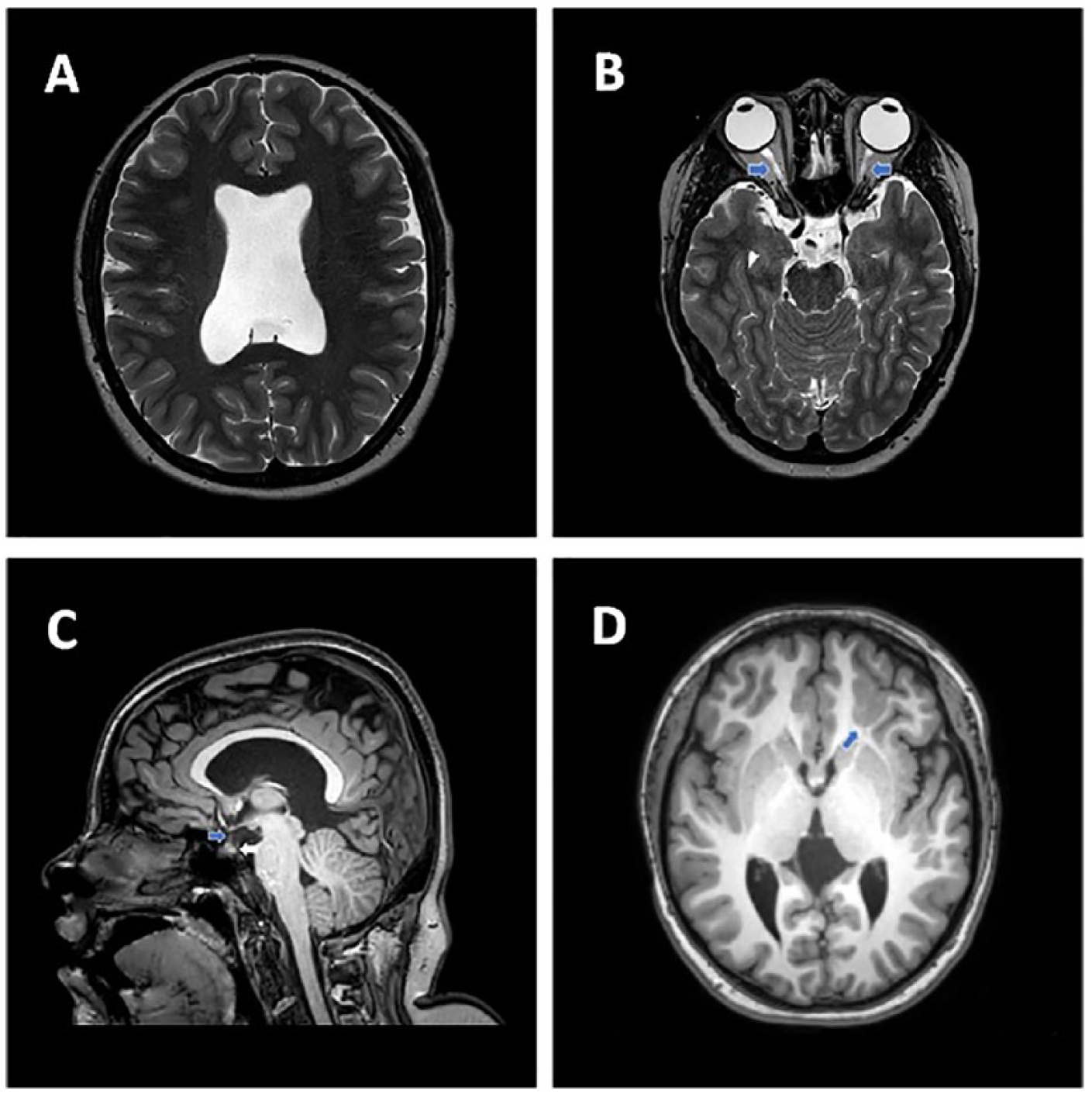
(A) Axial T2 weighted fat saturated image at the level of the lateral ventricles shows the absence of the septum pellucidum. (B) Axial T2 weighted fat saturated image at the level of the orbits shows diminutive optic nerves bilaterally (blue arrows). (C) Sagittal T1 weighted image at the level of the corpus callosum shows a small pituitary stalk (blue arrow) without visualization of a normal posterior pituitary bright spot (white arrow) suggesting posterior pituitary ectopia. (D) Axial T1 weighted image at the level of the basal ganglia shows subcortical grey matter heterotopia and polymicrogyria (blue arrow) within the left frontal lobe.
SOD is generally described as sporadic in frequency and of unclear etiology. 11 Diagnosis is made on clinical grounds as genetic evaluations to date have not yet elucidated the underlying genetic mechanisms responsible for SOD. Evaluations of genes controlling brain morphogenesis have shown mutations in the HESX1 and SOX2 genes in some SOD individuals. 12 Other closely related syndromes (ONH, CPHD) have demonstrated mutations in related genes involved in the ontogenic cascade of forebrain development, some of which (LHX3, HESX1) are associated with hearing loss.7,9 SOX2 and LHX3 have also been identified in the developing inner ear and may represent another potential connection between SOD and sensorineural hearing loss. 13 Enthusiasm for a genetic mechanism is tempered by the relative rarity of these mutations in the SOD population. A recent review of 850 SOD subjects identified HESX1 in less than 1% of cases. 14
Vascular disruption has also been proposed as a potential causal mechanism of SOD, which accounts for the lack of universal genetic/embryologic findings. Lubinsky 15 hypothesized that a vascular disruption sequence involving the anterior cerebral artery was a more likely etiological explanation for SOD than a genetic or embryologic explanation. He developed this theory based on the different embryological phases and genetic origins of the involved neural structures. 15 An association with gastroschisis, another proposed vascular disruption disorder, has also been presented as additional evidence of a potential underlying vascular source for SOD. 16 Proponents of this theory of cause also cite that the 3 abnormalities noted in SOD are found within the vascular supply of a common artery. 11 While this theory does not explain why hearing loss was noted in nearly 11% of our SOD cohort, these preliminary findings suggest further investigation is required to elucidate the potential in utero mechanisms for developing SOD.
Atresia of the external auditory canal is an uncommon congenital finding related to a failure of the external auditory canal to canalize during development. While atresia is associated with craniofacial syndromes such as Crouzon and Treacher Collins, most cases are unilateral and thought to be sporadic in etiology. 17 To our knowledge, atresia and/or microtia have not been previously reported with SOD. Future reviews of larger SOD populations are needed to determine whether a subpopulation of SOD patients with atresia and/or microtia exists.
The identification of cochlear nerve deficiency as a source for SNHL in SOD is also, to our knowledge, a new finding. While optic nerve hypoplasia is commonly noted in SOD, other cranial nerve deficiencies are not commonly reported in this population. With the introduction of universal newborn hearing screening programs and improving MRI capabilities, cochlear nerve deficiency has been increasingly identified as a source for congenital sensorineural hearing loss. A pediatric study reviewing MRI findings noted cochlear nerve deficiency in 18% of ears with hearing loss. 18 Interestingly, a recent review by Clemmens et al 19 identified ophthalmologic abnormalities in 67% of patients with cochlear nerve deficiency. Although the genetics of cochlear nerve deficiency are unknown, these results are similar to other reviews of pediatric SHNL demonstrating ocular findings in 22% to 32% of children. 20 While these studies suggest an association with ophthalmologic and otic abnormalities, further investigation is required to determine the prevalence of cochlear nerve deficiency in SOD.
Over half of SOD children with hearing loss in this study required tympanostomy tubes, suggesting the need for close monitoring of this population for otitis and middle ear effusion. Although the data did not suggest any difference in the natural history of otitis in children with SOD, the effects of Eustachian tube dysfunction can make proper amplification more difficult to achieve. As with all children with underlying hearing loss, SOD patients with hearing loss require increased vigilance for otitis and proactive treatment with tympanostomy tubes when medically appropriate.
Although limited by its retrospective design and small number of SOD patients, this study suggests an underappreciation for the prevalence of hearing loss associated with SOD. Children diagnosed with SOD failing newborn hearing evaluations should prompt further evaluation with dedicated temporal bone imaging to assess otic and cochlear structural integrity. Given the multiple sensory deficits associated with SOD hearing loss patients, aggressive early intervention of concomitant otitis is also recommended. Despite developmental delays associated with SOD, amplification was successfully implemented in the vast majority of these patients. Given the lack of cochleovestibular malformations noted in this SOD cohort, cochlear implantation is also a viable option for appropriate candidates. Physicians of all disciplines caring for SOD patients must maintain a high index of suspicion for potential hearing loss.
Conclusion
Physicians caring for pediatric SOD patients should remain vigilant for the possibility of hearing loss in this population. This study identified significant variability in type and severity of hearing loss, with bilateral SNHL the most common presentation. This study is, to our knowledge, the first to examine the association of hearing loss with SOD. It has also identified aural atresia and cochlear nerve deficiency associated with SOD hearing loss patients, underscoring the variable pathogenic mechanisms involved with hearing loss in this disorder. Close monitoring and prompt treatment of otitis to prevent additional conductive hearing loss is also important given the other sensory deficits associated with SOD. Early identification and intervention are recommended to limit the impact of hearing loss on SOD children.
Footnotes
Acknowledgements
Elyse Handley and Jordyn Dinwiddie for assistance in preparation and submission of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.