
Abstract
Background:
Recommendations regarding head and neck paragangliomas (HNPGL) have undergone a fundamental reorientation in the last decade as a result of increased understanding of the genetic and pathophysiologic basis of these disorders.
Objective:
We aim to provide an overview of HNPGL and recent discoveries regarding their molecular genetics, along with updated recommendations on workup, treatment, and surveillance, and their implications for otolaryngologists treating patients with these disorders.
Results:
SDHx susceptibility gene mutations, encoding subunits of the enzyme succinate dehydrogenase (SDH), give rise to the Hereditary Pheochromocytoma/Paraganglioma Syndromes. SDHA, SDHB, SDHC, SDHD, and SDHAF2 mutations each result in unique phenotypes with distinct penetrance and risk for variable tumor development as well as metastasis. Genetic and biochemical testing is recommended for every patient with HNPGL. Multifocal disease should be managed in multi-disciplinary fashion. Patients with SDHx mutations require frequent biochemical screening and whole-body imaging, as well as lifelong follow-up with an expert in hereditary pheochromocytoma and paraganglioma syndromes.
Conclusion:
Otolaryngologists are likely to encounter patients with HNPGL. Keeping abreast of the latest recommendations, especially regarding genetic testing, workup for additional tumors, multi-disciplinary approach to care, and need for lifelong surveillance, will help otolaryngologists appropriately care for these patients.
Keywords
Introduction
Otolaryngologists are well-trained to treat head and neck paragangliomas (HNPGLs), but several recent developments in molecular genetics and associated hereditary syndromes have led to a fundamental reorientation in recommended workup and long-term monitoring for patients with these tumors. Here, we provide an overview of HNPGLs, as well as an update on the molecular genetics, workup, and long-term surveillance recommendations. We aim to provide information that every otolaryngologist treating these disorders should know.
Incidence, Pathology, and Physiology
Paragangliomas (PGLs) are neuroendocrine tumors derived from neural crest cells that arise in extra-adrenal autonomic paraganglia, both sympathetic and parasympathetic; pheochromocytomas (PHEO) are closely related and arise from the adrenal medulla. HNPGLs comprise 65% to 70% of PGL (excluding PHEO) and account for 0.6% of head and neck tumors, with an overall incidence of 1 in 30 000 to 1 00 000.1-3
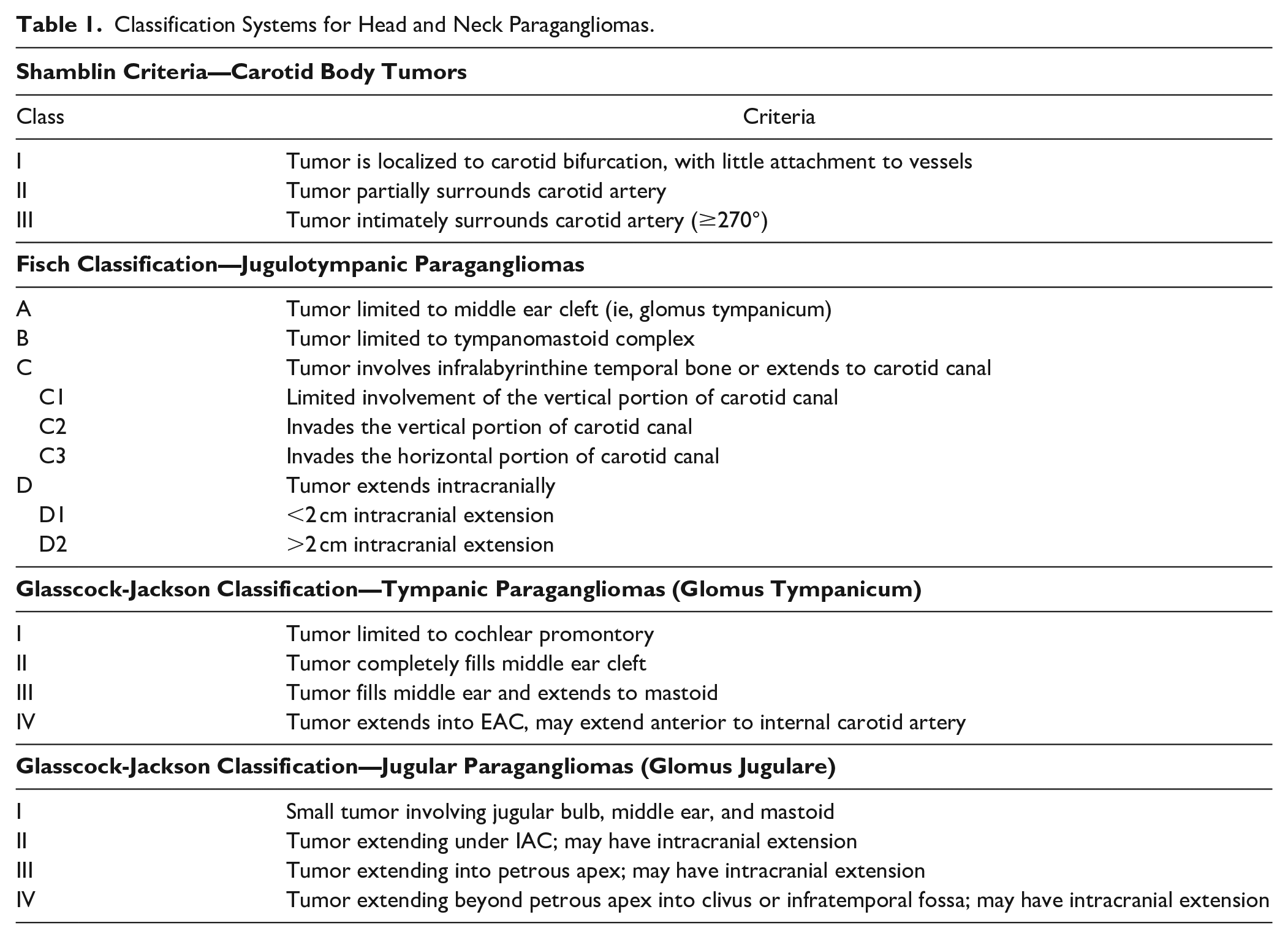
HNPGLs arise predominantly from the parasympathetic ganglia of the glossopharyngeal and vagus nerves within the skull base or upper neck. Carotid body tumors (CBT) are the most common HNPGL, constituting 50% to 60% of cases,3,4 and are classified according to Shamblin’s criteria 5 (Table 1). Jugulotympanic PGLs (JT PGL) arise from the jugular bulb (glomus jugulare) or from Arnold’s or Jacobson’s nerves within the middle ear (glomus tympanicum), and comprise 20% to 30% of HNPGLs. Though still popularly used, the term “glomus” is a historical misnomer arising from confusion with true glomus tumors derived from thermoregulatory myoarterial structures, and is no longer accepted pathologic nomenclature. 6 JT PGLs are characterized according to either Fisch 7 or Glasscock-Jackson 8 classifications (Table 1). Vagal PGLs account for 5% to 10% of HNPGLs,4,9 with even fewer arising from the larynx, trachea, thyroid, nasal cavity, and orbit. 10
Classification Systems for Head and Neck Paragangliomas.
Pathologists play a key role in diagnosing PGLs. On gross examination, these encapsulated tumors are firm and rubbery, and often ovoid in shape.3,4 Microscopically they are constituted by nests of chief cells known as Zellballen, though this classic architecture is not universally observed.6,11 Cytologically they demonstrate the clear cytoplasm characteristic of neuroendocrine tumors. 4 Immunostaining of PGLs is positive for neuroendocrine markers including chromogranin, synaptophysin, neuron-specific enolase, and S-100, while being negative for cytokeratin.3,4
Production of metanephrines (metanephrine and normetanephrine) and catecholamines (epinephrine, norepinephrine, and dopamine) is characteristic of PHEOs as well as sympathetic PGLs, which are most commonly found along the thoracic, abdominal, and pelvic paravertebral axis. 12 HNPGLs, however, rarely secrete catecholamines (<4%)1,13-15 due to their predominantly parasympathetic origin. Defining malignancy in HNPGL is a controversial issue due to the difficulty of distinguishing true metastasis (paraganglionic cells in non-neuroendocrine tissue distant from the primary tumor) from multicentric primary neoplasms. Currently, the World Health Organization (WHO) classifies PGL as a tumor of indeterminate biology. 16 They are not categorized according to the typical benign vs malignant paradigm but fall along a spectrum of metastatic potential. 17 Nevertheless, outside of this pathologic characterization, HNPGLs generally exhibit clinically benign behavior; recent analysis demonstrates rate of definitive metastasis to be <4%. 18
Clinical Presentation and Management
The presentation of symptomatic patients with HNPGL differs based on the location and physiology. Symptoms, if any, are most often due to local mass effect. CBTs and vagal PGLs may present with a neck mass, cough, hoarseness, or dysphagia; JT PGLs with pulsatile tinnitus, hearing loss, or other lower cranial nerve deficits. 19 Actively secreting tumors, though uncommon in HNPGL, precipitate symptoms of catecholamine excess: hypertension, headache, diaphoresis, palpitations, orthostasis, or anxiety. 1 With escalating medical imaging use in the modern era, 20 HNPGLs also increasingly present as an incidental finding.1,21
Management options for HNPGLs include observation, surgery, and radiation. In addition to alleviating current symptoms, the main goals of treatment include optimal balancing of tumor-induced and treatment-induced complications. Several factors influence management decisions including patient age, comorbidities, cranial nerve status, tumor size, and tumor location(s);19,22-24 symptoms specific to each type of HNPGL (eg, pulsatile tinnitus in glomus tympanicum) may also guide treatment decisions.19,24 Because HNPGLs are typically very slow growing or even stable in size21,25 the natural time course factors heavily into the decision-making process regarding observation versus intervention of any sort. Due to this slow growth rate as well as the benign nature of most HNPGLs, observation is a reasonable initial option for many patients.26,27 This strategy is referred to by some as “wait-and-scan,” 28 though we prefer the term “active observation” to decrease patient anxiety elicited by the word “wait”, and to emphasize the continuing monitoring required. Observation is often indicated as an initial method to help delineate growth rate, especially in asymptomatic patients for whom therapy poses significant risk for iatrogenic injury, and in patients of advanced age or those with pre-existing contralateral cranial nerve deficits.19,21,26,27 A conservative approach is increasingly being recommended for glomus jugulare in particular due to the risk of iatrogenic lower cranial nerve deficit with resection.29-31
Of course, avoiding treatment-induced complications exposes patients to the risk of tumor-induced sequelae, and there is always concern for progression of cranial nerve deficits during the period of observation, especially in young patients. CBTs often demonstrate growth over long follow-up time periods, 32 and large tumors (Shamblin III) result in a high rate of cranial neuropathy (12-79%). 32 A retrospective review of the observation approach in HNPGL demonstrated new complications, mostly cranial neuropathies, in 10% of patients with CBT, 14% with vagal PGL, and 15% with JT PGL during observation (median follow-up 5.3 years), many of which occurred in non-growing tumors. 25 Other retrospective studies show that observation over 3 years resulted in new lower cranial nerve deficits in 30% of patients with JT PGLs, 28 and, for those with glomus jugulare specifically, cranial nerve VII, X, XI, and XII deficits in 6%, 13%, 13%, and 19% (median follow-up 7.2 years). 26
Surgery is the treatment of choice for all PHEO and secretory PGL, and has traditionally been the mainstay of treatment for HNPGL also. Prior to the mid-20th century, CBT resection was associated with high rates of postoperative cranial neuropathy, stroke, and death. 33 Surgery for JT PGL had similarly grim outcomes, particularly regarding lower cranial nerve deficits, until a few decades ago. 34 Patients with larger tumors fare worse—the rate of new cranial neuropathies after attempted gross total resection of Fisch class C or D jugulotympanic tumors ranges from 39% up to 81%,31,35-38 and the rate of serious complications (cranial neuropathy or stroke) after resection of Shamblin III CBTs ranges from 33% to 50%.39-41 Conversely, smaller tumors are more amenable to safe and successful resection: postoperative cranial neuropathy occurs in 6% of Shamblin class I 39 and 14% to 18% of Shamblin class II CBTs,39,42 and 8% to 13% of Fisch class C JT PGLs. 36 With recent advances in neuroradiology, neuroanesthesia, and microsurgery, surgical resection is now an even safer and more feasible option, including some cases formerly deemed inoperable. Preoperative embolization is commonly used for HNPGL as some authors demonstrate decreased blood loss and operative time in CBT resection,43-45 though others find no improvement in outcomes.46,47 Embolization may rarely disrupt cranial nerve blood supply, likely due to underlying vascular anatomic variants or the use of the liquid embolization agent ethylene vinyl alcohol (Onyx®). 48 Subtotal resection for JT PGL has been increasingly utilized to decrease morbidity in select patients, including those of advanced age, larger tumor size, or with bothersome symptoms including hearing loss or tinnitus. 31
Radiotherapy is another treatment modality option for HNPGLs, used as either primary or adjuvant therapy. Radiotherapy is used primarily for JT PGL, though in some circumstances it may be utilized for CBTs or other HNPGL. Similar to surgery, initial treatments with ionizing (orthovoltage) radiation in the mid-20th century produced an unacceptably high rate of complications; 49 more modern megavoltage external beam radiation achieved improvements in tumor control with fewer complications.49-51 Over the past few decades, stereotactic radiosurgery (SRS) has emerged as a viable option for JT PGLs. Primary treatment with SRS produces excellent short- and intermediate-term tumor control (92-95%);52,53 the risk of cranial neuropathy is 3% to 12%.22,54 One recent retrospective study of glomus jugulare tumors confirms excellent 5-year (98%) and 10-year (94%) tumor control, but demonstrates a drop-off in tumor control to 74% at 15-year follow-up. 54 As JT PGLs are rare and the data is retrospective, detailed comparisons of surgical and radiation outcomes are challenging, and are beyond the scope of this review.
Hereditary Syndromes and Genetics
PGLs may be sporadic or inherited. The hereditary syndromes and associated genetic mutations classically associated with PGL and PHEO formation include Multiple Endocrine Neoplasia type 2 (RET mutation), Neurofibromatosis type 1 (NF1 mutation), and von Hippel Lindau Syndrome (VHL mutation). 13 These syndromes are usually associated with adrenal PHEO and initially were the only known inherited syndromes associated with PHEO; therefore, it was believed 10% were hereditary. Our understanding has significantly changed over the past two decades since the discovery of several new genes associated with hereditary PHEO/PGL. PHEO/PGL now has the highest known hereditary predisposition of any solid tumor type—up to 40% of cases—surpassing medullary thyroid carcinoma. 55
A familial disposition for these tumors was first postulated in 1933 by Chase. 56 Subsequently, “familial paraganglioma syndromes” were determined to display autosomal dominant inheritance with variable penetrance, characterized by a predisposition to PGLs, PHEO, renal cell cancers (RCC), gastrointestinal stromal tumors (GIST), and pituitary adenomas. It was not until 2000, however, that the genetic basis for hereditary PGL syndromes was discovered, when Baysal et al implicated a mutation in the gene encoding succinate dehydrogenase (SDH) in the development of CBTs. 57 This enzyme, utilized in both the tricyclic acid cycle of oxidative phosphorylation and in the mitochondrial electron transport chain, has four subunits, A-D: SDHA, SDHB, SDHC, and SDHD, as well as the flavination/assembly factor SDHAF2. 57 Without SDH activity, succinate cannot be oxidized to fumarate and accumulates within the cell, inhibiting prolyl hydroxylases and stabilizing the HIFα transcription factor, promoting angiogenesis and rendering the cell less susceptible to hypoxia.13,15 Mutations in any of the genes encoding these SDH subunit proteins are associated with a genetic predisposition for developing PGLs and PHEO. Subsequently, additional susceptibility genes have been implicated in the formation of PHEO/PGL—principally MAX and TMEM127, and also in rare cases possibly EGLN1, EGLN2, KIF1β, IDH1, HIF2A, MDH2, FH, SCL25A11, and DNMT3A.14,58
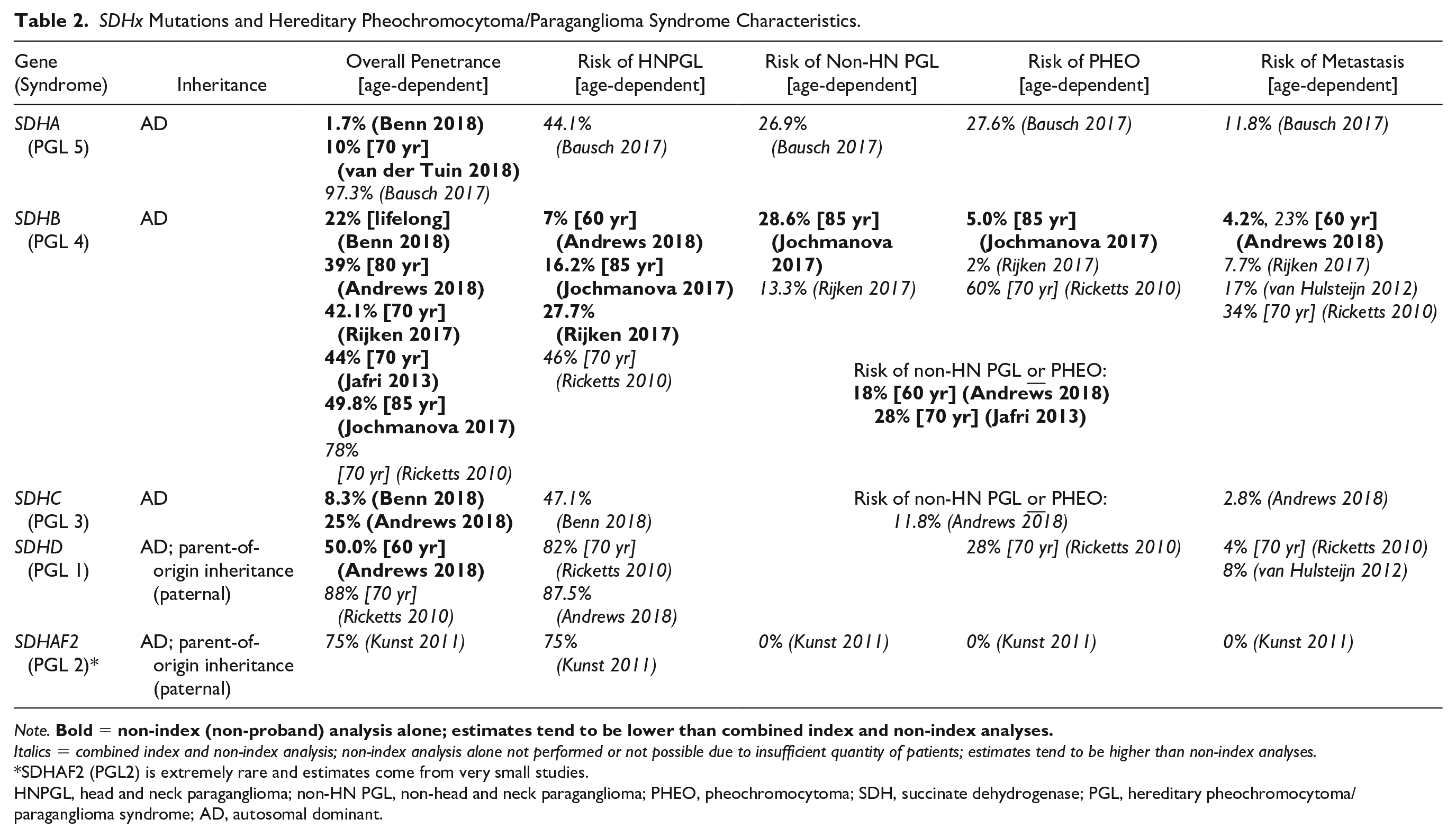
Since initial discovery of SDHx as susceptibility genes for PHEO/PGL, genotype-phenotype correlations for each gene have been established. The SDHx genes cause the Hereditary PGL/PHEO Syndromes, outlined in Table 2; of note, this nomenclature replaces the previously existing term “Familial PGL Syndromes.” These syndromes are transmitted in autosomal dominant fashion, with age-dependent and incomplete penetrance.59,60 In SDHD and SDHAF2 mutations, inheritance displays evidence of parent-of-origin preference, wherein only paternally inherited mutated genes lead to tumor formation. 59
SDHx Mutations and Hereditary Pheochromocytoma/Paraganglioma Syndrome Characteristics.
Note.
Italics = combined index and non-index analysis; non-index analysis alone not performed or not possible due to insufficient quantity of patients; estimates tend to be higher than non-index analyses.
SDHAF2 (PGL2) is extremely rare and estimates come from very small studies.
HNPGL, head and neck paraganglioma; non-HN PGL, non-head and neck paraganglioma; PHEO, pheochromocytoma; SDH, succinate dehydrogenase; PGL, hereditary pheochromocytoma/paraganglioma syndrome; AD, autosomal dominant.
SDHA pathogenic variants (PGL 5) leading to PHEO/PGL are rare as the penetrance is quite low, but if a patient develops an SDHA-associated PHEO/PGL, the risk of metastatic disease may be high.61-63 SDHB related Hereditary PGL/PHEO Syndrome (PGL 4) is characterized by a high prevalence of sympathetic PGL, followed by PHEO and then HNPGL; although this syndrome was initially thought to result in significantly higher risk of metastatic disease, more recent studies have moderated this estimation, although it remains higher than for the other subunits.18,59,63-67 SDHC related Hereditary PGL/PHEO Syndrome (PGL 3) displays a high prevalence of HNPGL with low but present risk of sympathetic PGL, PHEO, and metastasis.63,64 SDHD related Hereditary PGL/PHEO Syndrome (PGL 1), the most common SDHx-related syndrome, is marked by multifocal HNPGL and elevated risk of developing sympathetic PGL and/or PHEO, with low potential for metastasis.64,66,68 SDHAF2 pathogenic variant (PGL 2) carriers are rare and present with either single or multifocal HNPGL without significant risk of sympathetic PGL, PHEO, or metastasis. 62
Workup
Given the high rate of inherited syndromes among patients with HNPGL, workup of a head or neck mass suspicious for PGL should include biochemical testing to screen for additional primary PHEO/PGL in addition to the rare secretory HNPGL. Biochemical screening should include plasma free metanephrines, or 24-hour urine fractionated metanephrines and catecholamines.12,69 Several factors may lead to falsely elevated results, including sympathomimetic activities such as cigarette smoking as well as several drugs: caffeine, acetaminophen, some beta- and alpha-adrenergic blockers, and many classes of antidepressants such as tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, and monoamine oxidase inhibitors. 13 Plasma free metanephrine samples should ideally be collected from an indwelling venous catheter after 30 minutes in the supine position, to minimize confounding spikes in endogenous catecholamine release.12,15 This is not always practical, and if the results are normal when the patient is sitting up, no further testing is required. Although the sensitivity of plasma free (99%) and urine fractionated (97%) metanephrines is high, specificity is lower (89% and 69%, respectively), and false positives are common. 69 Biochemical management of PHEO and secretory PGL, including the rare secretory HNPGL, involves a preoperative alpha-adrenergic blockade to avoid hypertensive crisis from surge of catecholamines during anesthesia induction and surgical resection of the tumor. 12
Site-specific imaging, typically computed tomography (CT) or magnetic resonance imaging (MRI), is often necessary for growth monitoring or surgical planning for HNPGL. In addition, if biochemical testing shows elevated plasma free metanephrines, whole-body CT or MRI (skull base to pelvis) with and without contrast is recommended to screen for additional primary PHEO/PGL tumors. This testing has a sensitivity of 88% and specificity of 95% for identifying additional tumors. 70 If specifically evaluating for metastatic disease or small multifocal lesions, functional imaging may be used, though incurring higher radiation exposure: fluorodeoxyglucose positron emission tomography (PET)/CT, 68-Ga DOTATATE PET/CT, or 123I–MIBG. 59 The 68-Ga DOTATATE scan has the highest detection rate.71,72
Genetic testing is recommended for all patients presenting with HNPGL. 55 The lowering costs of gene panel testing and widespread adoption of coverage by insurance companies, combined with the ineffectiveness of family history screening for predicting syndrome risk (due to overall low penetrance), has led to this recommendation.
Multi-Disciplinary Approach
For patients with a single HNPGL and negative genetic testing, balancing the risks of surgery, radiotherapy, and observation (as discussed in above sections) has not significantly changed with the advent of the recent molecular genetics data presented here. However, if additional tumors (multiple HNPGLs or PHEO/PGL in another location) are discovered after appropriate workup of a HNPGL, multi-disciplinary discussion regarding treatment should include an otolaryngologist and endocrinologist or medical geneticist familiar with the hereditary genetics of these tumors. In almost every case, any non-HNPGL should be removed prior to resection of any HNPGL given the risk for hypertensive crisis with catecholamine secretion or storage in sympathetic-derived PHEO/PGL. In patients with a single HNPGL and known SDHx susceptibility gene mutation, careful consideration should be given to whether primary surgery or radiotherapy is in the best interests of the patient; similar to multidisciplinary decision-making for patients with NF2, potential future treatments for other tumors should be taken into account when making this decision. Significant knowledge gaps remain concerning best practice with respect to balancing risk of current treatment with future treatment risk. Patients with metastatic disease may be candidates for chemotherapy, clinical trials, 131I–MIBG therapy, or external beam radiation; in such situations, consultation with medical oncology, endocrinology, nuclear medicine, and/or radiation oncology is indicated. 59
Surveillance
If a patient has pathogenic variants in any of the SDHx susceptibility genes predisposing to development of PGL or PHEO, annual biochemical screening along with whole-body MRI (skull base to pelvis) every 2 to 3 years are recommended to identify additional asymptomatic PGL or PHEO or other tumors related to these syndromes (ie, RCC or GIST).14,58 The patient should be followed for life by an expert in the field, such as an endocrinologist, nephrologist, or medical geneticist, due to the lifelong risk of tumor development. Figure 1 outlines the recommended approach to workup and surveillance in patients with HNPGL.
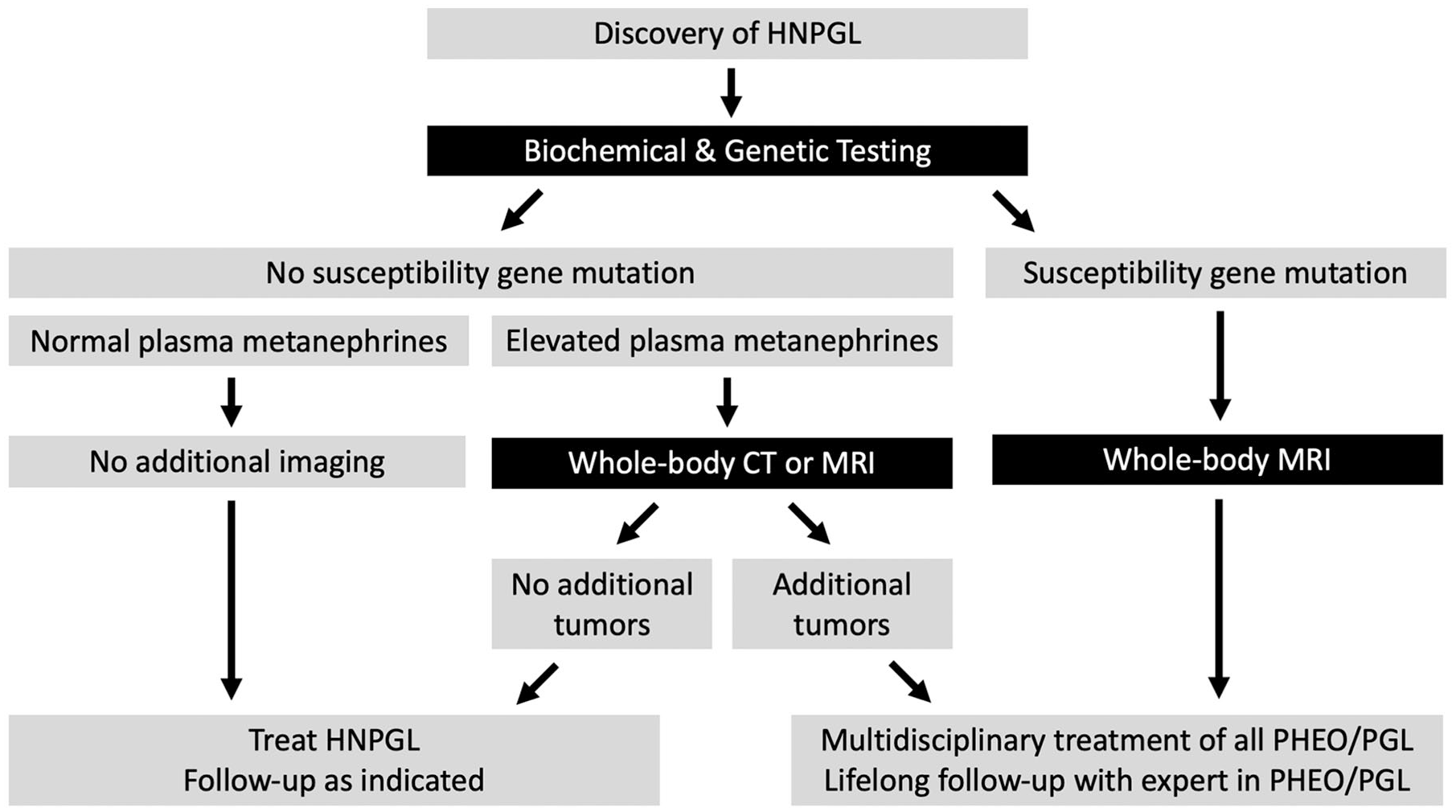
Recommended approach to workup and surveillance for patients with HNPGL.
Conclusion
HNPGL comprise the majority of extra-adrenal PGL, and otolaryngologists are likely to encounter patients with such disease. Keeping abreast of the latest recommendations, especially regarding genetic testing, workup for additional tumors, multi-disciplinary approach to care, and need for lifelong surveillance, will help otolaryngologists appropriately care for these patients.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support for LF from the American Cancer Society Mentored Research Scholar Grant MRSG-15-063-01-TBG. SG receives grant support from the NIH/NIDCD for work unrelated to this project and is on the scientific advisory boards for Applied Genetic Technologies Corporation and Roche as well as the data monitoring committees for Pipeline Therapeutics and the Cystic Fibrosis Foundation. He has also received an honorarium from Decibel Therapeutics for consulting and is a consultant for Cochlear Corporation and Sirocco Therapeutics. In addition, he is on the advisory board for the Cystic Fibrosis Foundation and receives research support without personal financial remuneration from Med-El Corporation.