
Abstract
We demonstrate deep ultraviolet (UV) photoacoustic spectroscopy (PAS) of trace explosives using a sensitive microphone at meter standoff distances. We directly detect 10 µg/cm2 of pentaerythritol tetranitrate (PETN), 2,4,6-trinitrotoluene (TNT), and ammonium nitrate (AN) with 1 s accumulations from a 3 m standoff distance. Large PAS signals for standoff detection are achieved by exciting into the absorption bands of the explosives with a 213 nm laser. We also investigate the impact of the deep UV photochemistry of AN on the PAS signal strength and stability. We find that production of gaseous species during photolysis of AN enhances the PAS signal strength. This deep UV photochemistry can, however, limit the PAS signal lifetimes when detecting trace quantities.
Keywords
Introduction
Explosives pose a serious threat to the safety of civilians and military personnel. Early detection of explosives is essential for mitigating these threats and for tracking attribution. 1 We can provide early warning for these threats by screening for the presence of explosives on contaminated surfaces. This is possible because individuals handling explosives often transfer explosive particulates to surfaces they encounter.1–4
Current methodologies to screen for explosives include canine olfaction, ion mobility spectrometry, and colorimetric and fluorometric assays;1,3 unfortunately, these approaches require close contact with the contaminated surfaces. Approaching a suspicious object to screen for explosives is risky. Consequently, there is a need to develop alternative, standoff methodologies for trace explosive detection.
Laser-based spectroscopies,3,5,6 such as Raman spectroscopy, laser-induced breakdown spectroscopy (LIBS), and photoacoustic spectroscopy (PAS), have gained attention for the standoff detection of explosives. Raman spectroscopies provide vibrational fingerprints for identifying explosives, but these methods often have weak signals requiring long accumulation times. These methods can also have strong background interferences such as fluorescence. Laser-induced breakdown spectroscopy provides strong signals, short accumulation times, and long standoff distances, but the LIBS experimental design and analyte identification process can be complicated.
In this work, we focus on developing PAS because of its simplicity and ease of implementation for non-specialists. Photoacoustic spectroscopy has fast response times and shows weak to nonexistent substrate signals. Moreover, the photoacoustic signal decays by 1/r with detection distance, compared with 1/r2 for other spectroscopic techniques.7,8
Photoacoustic spectroscopy is based on the heating of a sample by the absorption of light. When solids absorb radiation, they transfer heat to the surrounding air. The resulting increase in air temperature and subsequent expansion of the air drives an acoustic pulse, often referred to as the thermal-piston effect.9–11 By using a modulated light source, such as a pulsed laser, the photoacoustic signals can be detected with a sensitive microphone.
The evolution of heat at the solid-air interface relies on sample properties including the absorption coefficient and the thermal diffusivity.7,9–11 The absorption coefficient determines the heated depth and temperature rise of the solid sample. The thermal diffusivity (α = K/pCp) determines the heat flow through the system, where K is the thermal conductivity, Cp is the heat capacity, and p is the solid density. Typically, larger absorption coefficients and thermal conductivities, and smaller heat capacities lead to larger PAS signals. Contributions from induced mechanical vibrations of solids are considered minimal due to their small thermal expansion coefficients relative to gases. The mechanical vibrations of solids, however, have been shown to contribute to the PAS signal in certain cases. 12
Previous studies of standoff PAS have explored the use quantum cascade lasers (QCLs) in the infrared (IR) to detect military grade explosives such as 2,6-dinitrotoluene (DNT), 2,4,6-trinitrotoluene (TNT), pentaerythritol tetranitrate (PETN), and 1,3,5-trinitroperhydro-1,3,5-triazine (RDX).13–16 These studies relied primarily on quartz crystal tuning forks (QCTFs) claiming detection limits between 100 ng/cm2 and 5 µg/cm2.14,16,17 Quantum cascade lasers are advantageous because of their wavelength tunability, but their use for PAS does not take advantage of the strong deep ultraviolet (UV) absorption bands of explosives. In contrast, a study by Wynn et al. examined the PAS response of TNT, RDX, and DNT using a laser Doppler vibrometer and 266 nm excitation. 18 They estimated a detection limit of 100 ng/cm2 at a 1 m standoff distance.
The peak absorptions for most explosives, however, are at λ < 260 nm.19,20 For example, PETN and HMDT (hexamethylene triperoxide diamine) have maximum absorptions at <190 nm, ammonium nitrate (AN) has a maximum absorption at ∼200 nm, and TNT, HMX (cyclotetramethylenetetranitramine), and RDX have maximum absorptions at ∼230 nm. By exciting into the absorption bands of explosives, we can achieve large PAS signals.
Many explosives also undergo photochemistry when irradiated with deep UV light.21–26 Large photoacoustic responses have been observed for photochemical reactions. 27 As we show here, this provides an additional avenue for signal detection. By using a microphone, we can exploit the photoacoustic signals produced during the formation of gaseous species for standoff detection. This contrasts with previous QCTF-based approaches, which indirectly produce a photoacoustic response by focusing the IR scattered light from the explosive target onto a tuning fork.14,16,17
In this investigation, we developed an instrument for the standoff detection of trace explosives by using deep UV PAS with a sensitive microphone. We use a laser wavelength of 213 nm, which is near to the peak absorptions of most explosives to induce a PAS response. We studied TNT, PETN, and AN because they have absorption maxima in the deep UV around our laser excitation wavelength.19,20,23 We also investigate the influence of photochemistry on the PAS signal strength and stability. The deep UV PAS properties of explosives allow us to achieve large standoff distances with a straightforward instrument design and modest laser powers. Using deep UV PAS, we directly detect 10 µg/cm2 of PETN, TNT, and AN with 1 s accumulations at standoff distances of 3 m.
Methods
Sample Preparation
One-inch squares of PETN, AN, and TNT were ink-jet printed on aluminum substrates (ACT Test Panel Technologies) at loadings of 10, 100, and 250 µg/cm2. These samples were donated by the US Army Research Laboratory (ARL). Holthoff et al. describe their preparation. 28 Ink-jet printing is more reproducible than drop-casting because it provides control over the volume, location, and density of droplets deposited on the substrate. While ink-jet printing is an improvement over drop-casting, variability in sample coverage was still observed.
The nitrate salts investigated were AN (NH4NO3, J.T. Baker, purity 99.8%), sodium nitrate (NaNO3, Fisher Scientific, purity 99.8%), potassium nitrate (KNO3, J.T. Baker), and rubidium nitrate (RbNO3, Sigma Aldrich, purity 99.95%). Solid samples of each nitrate salt were prepared on plane microscope glass slides (Fisherbrand) by melting the granular analyte particles in an oven. After melting, the oven was turned off and the molten samples remained in the oven until solidification. After preparation, the samples were stored in a desiccator. Fresh solid samples were prepared for each experiment daily.
PAS Data Collection
Ink-jet printed PETN, AN, and TNT at loadings of 10, 100, and 250 µg/cm2 were investigated at 1, 2, and 3 m detection distances. 27 We used 60–80 mW of laser power measured at the output of a compact 213 nm neodymium-doped yttrium orthovanadate (Nd:YVO4) laser (15 ns pulse width) previously described. 23 The laser has a tunable repetition rate which was set to 20 kHz or 27 kHz. The laser beam was directed to the samples using a dielectric mirror (CVI Laser Optics) and was focused to a diameter of ∼1 mm.
For the trace explosive samples at each distance, 14 acquisitions were collected from two to five locations on the sample. Each acquisition was 1 s. The average and one standard deviation of the PAS signal were calculated for each explosive sample. Additional PAS responses over 30 s for PETN and AN at 3 m were measured for the 10 µg/cm2 samples. The background signal for each experiment was determined by averaging the signal collected by the microphone from over 100 acquisitions with the laser beam blocked.
We also investigated the interference signal from substrates such as aluminum, steel, and glass using the same 213 nm laser powers as for the trace explosive samples. The signal for each substrate was accumulated for 1 s for a total of 20–30 acquisitions. For Al, a blank portion of the Al substrate of the AN 10 µg/cm2 sample was irradiated at 3 m. For steel, a steel plate (303 Stainless Steel, McMaster Carr, 3366T228) was irradiated at 1 m. The steel plate was polished with sand paper (Norton P800), washed with soap, and rinsed with water prior to irradiation. For glass, a microscope glass slide (Fisherbrand) was irradiated at 1 m. The background signal was collected with the laser beam blocked under otherwise identical conditions for each substrate.
The deep UV PAS laser irradiance dependence studies for the nitrate salts were measured over a 1–80 mW power range for the 213 nm laser with a 20 kHz repetition rate and a laser spot diameter of 0.7 mm. The microphone was placed ∼2.5 cm from the sample at an angle of ∼30°. The average and one standard deviation of the PAS response were determined from 5–30 acquisitions. Laser power was simultaneously recorded with the photoacoustic response using a Thorlabs power meter (PM200).
The laser-induced temperatures of the nitrate salts were measured using a FLIR A325sc IR camera. For the steady state temperature studies of AN and NaNO3, the temperature was controlled using thermal heating tape and was recorded using the IR camera. The PAS signal from 30 acquisitions at each temperature for AN and NaNO3 was normalized for laser irradiance at 2–5 W/cm2.
Discussion
Deep UV Standoff PAS Instrument
Figure 1 shows a schematic of the instrument we developed for detecting the PAS of explosives. A compact 213 nm Nd:YVO4 laser was used to induce the PAS response.
23
This laser has a tunable repetition rate, which allows tuning of the PAS signal frequency. The PAS signals were collected with a sensitive microphone (Sennheiser MKH 8020, frequency response: 30 Hz–50 kHz, sensitivity: –30 dBV/PA, equivalent noise level: 10 dB(a)) with a Focusrite ISA One pre-amplifier at a gain of 50. To increase the standoff detection distance, we used a parabolic dish (Wildtronics, LLC) mounted on a tripod to collect and focus the PAS signals onto the microphone (Fig. 1b). The parabolic dish has a foam windscreen, which mitigates noise introduced by wind at speeds up to 4.5–5.4 m/s. A lock-in amplifier (Stanford Research) with a time constant between 300 ms and 1 s and a 12 dB filter was used to improve the signal-to-noise ratio. The reference input for the lock-in amplifier was from the function generator output of the laser. The output PAS signals were displayed on an oscilloscope (Tektronics) and exported to a computer for analysis. Data collection was semi-automated by using a custom-built Python program.
(a) Schematic of the deep UV photoacoustic instrument. A deep UV laser (λ = 213 nm) induces the PAS of trace explosives.
23
A parabolic dish (b) collects the sound and focuses it onto the microphone, which is amplified by a pre-amp. A lock-in amplifier is used to improve signal-to-noise. The signals are displayed on an oscilloscope prior to computer analysis.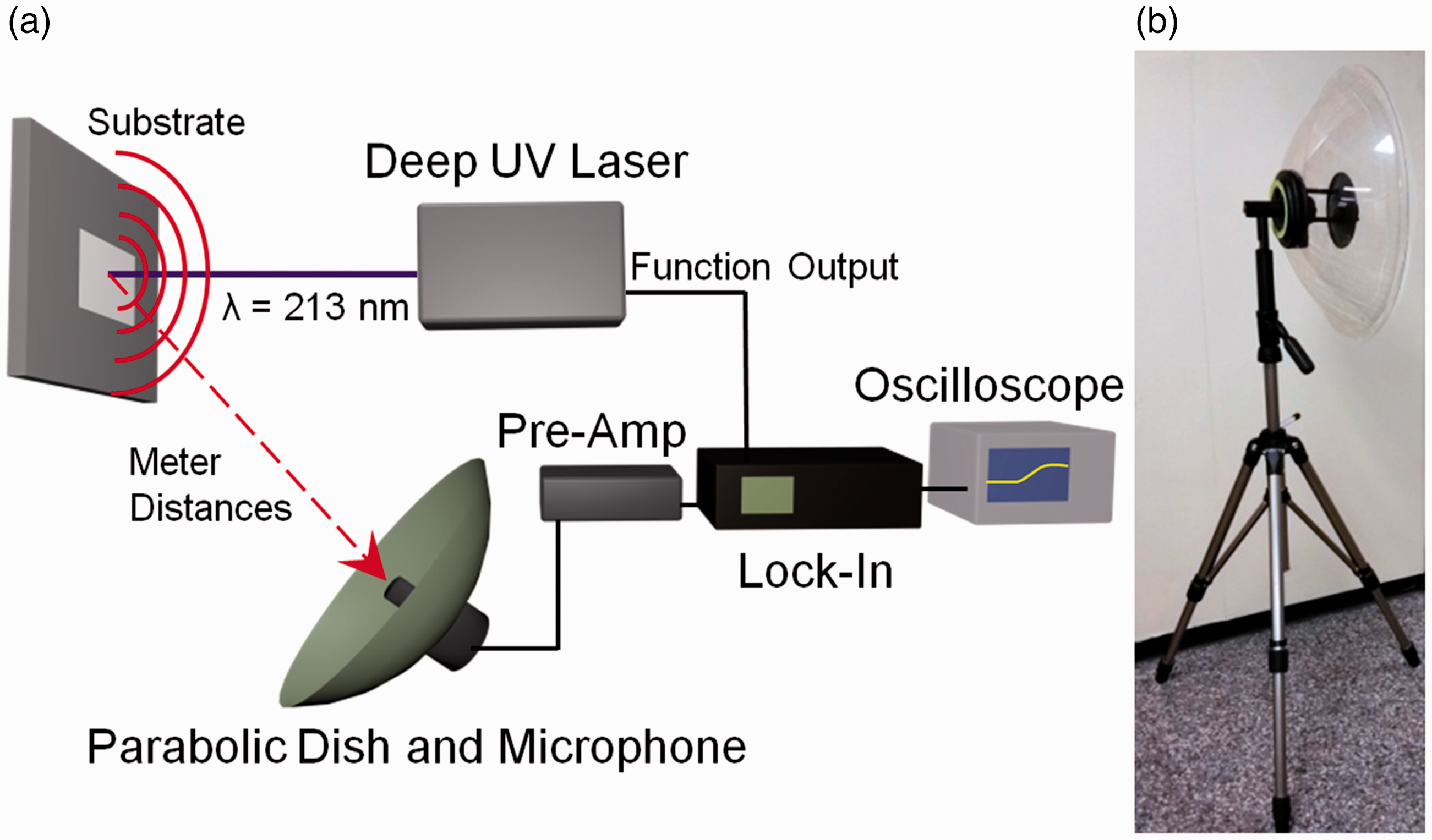
Background Signal Interference
For the deep UV PAS instrument, the primary approach for improving detection limits was mitigating the background signal interference. The background signal refers to signals collected by the microphone when the laser is not directed onto an explosive sample.
One source of interference in our experiments derives from mechanical acoustic generation centered in the laser cavity, which produces very weak signals at the same frequency as the PAS signal. Additional interferences presumably arise from electromagnetically excited acoustic noise and vibrations from other instrumentation and electronics in the laboratory. The variable repetition rate of the laser allows us to select measurement frequencies that minimize background signal interferences.
We also investigated potential substrate PAS interferences (see Supplemental Material Fig. S1). For this, we measured the PAS response produced by aluminum, steel, and glass substrates. We observed no significant difference between the baseline signal (laser beam blocked) and the laser directed onto blank aluminum, steel, or glass substrates at meter standoff distances.
For each experiment, we collected over 100 acquisitions to determine the average and standard deviation of the background signal. We used these results to define PAS signal thresholds that defined the explosive detection limits. In addition, we collected multiple acquisitions to determine the average and standard deviation of the PAS explosive signals. We used a t-test (unpaired, two tailed, unequal variances) to test that the mean PAS signal for the explosive differs from the background.
Standoff Detection of Trace Explosives
In Fig. 2, we show the PAS signal of TNT, PETN, and AN collected at standoff distances of 1, 2, and 3 m. At each distance, we measured three explosive loadings that were ink-jet printed on Al substrates: 250, 100, and 10 µg/cm2. We used 60–80 mW of laser power measured at the output of the compact 213 nm laser. With minimal optimization, we were able to detect the 250 and 100 µg/cm2 samples of TNT, PETN, and AN at all three detection distances, as well as the 10 µg/cm2 samples at the 1 and 2 m detection distances with a 20 kHz repetition rate.
Deep UV PAS signal for (a) TNT, (b) PETN, and (c) AN at 250 (red circle), 100 (blue diamond), and 10 (green square) µg/cm2 ink-jet printed samples on Al substrates measured at 1, 2, and 3 m. The average PAS signal is shown at each detection distance. The error bars represent the standard deviation of the signal collected under the same conditions. Average and standard deviation of the background signal (black) was calculated from over 100 acquisitions. The PAS signal was collected by using a 20 kHz repetition rate.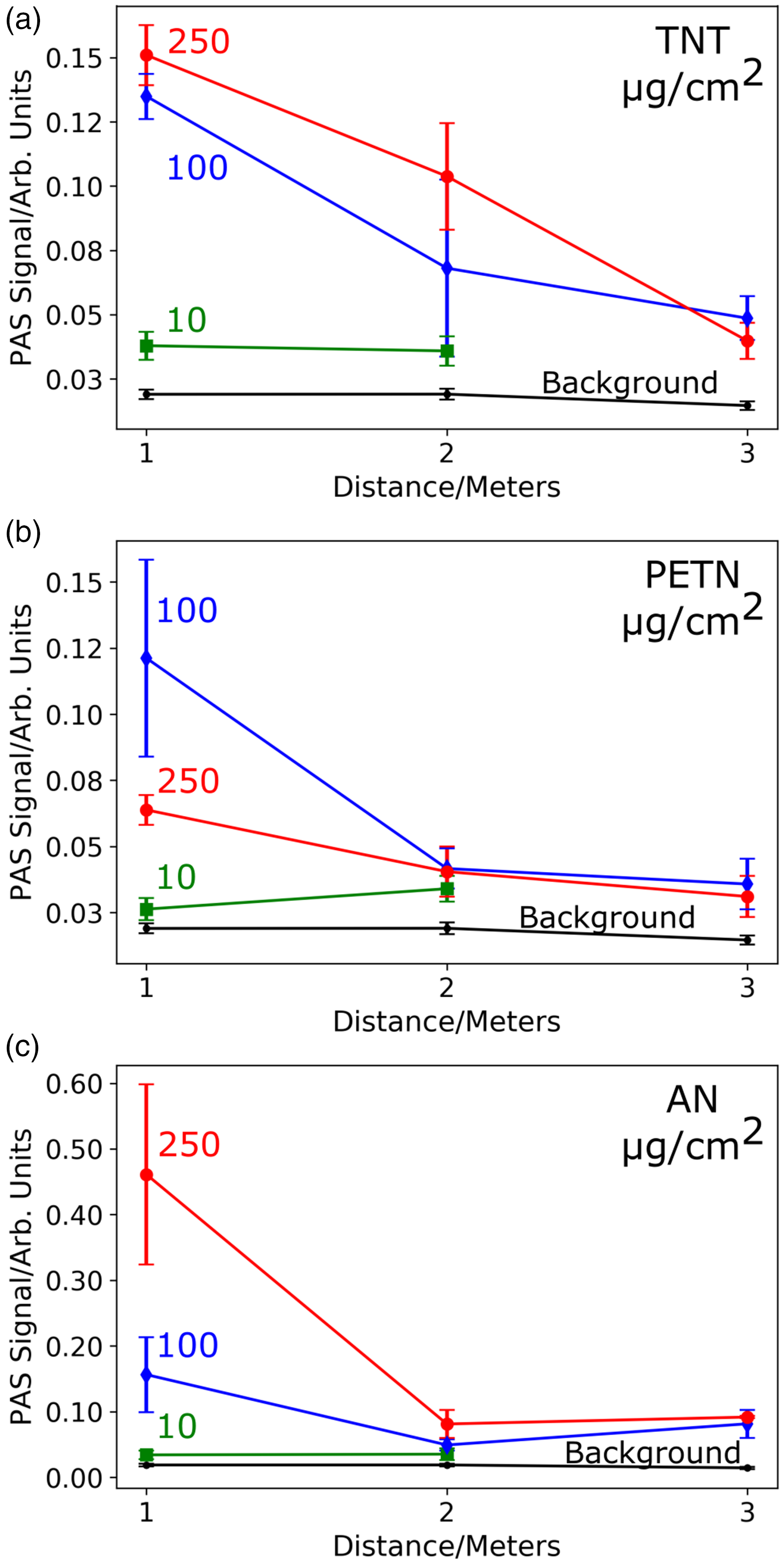
The detection of the 10 µg/cm2 explosive samples at 3 m was limited by background signal interference. To improve the detection limit, we investigated the laser repetition rate dependence of the PAS signal-to-noise (see Fig. S2). We found that a repetition rate of 27 kHz had less background signal interference and provided a factor of two to three better microphone response. By switching to 27 kHz, we were able to improve our limit of detection, achieving 3 m standoff detection of AN, PETN, and TNT at 10 µg/cm2, as shown in Fig. 3.
Deep UV PAS signal at 3 m for 10 µg/cm2 of TNT, PETN, and AN ink-jet printed on Al substrates. The average PAS signal is shown for each explosive. The error bars represent the standard deviation of signal collected under the same conditions. The PAS signal was collected at a 27 kHz repetition rate.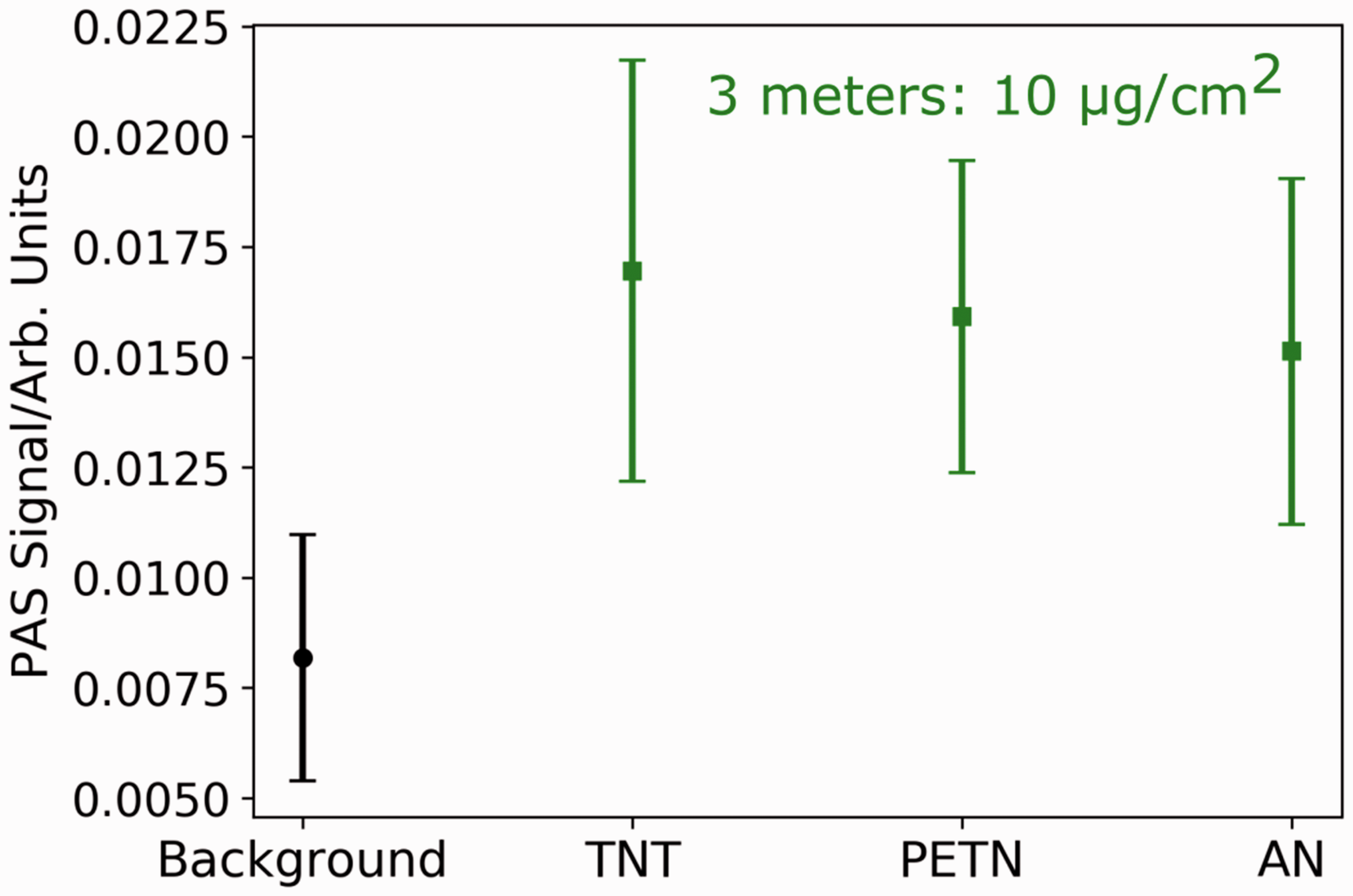
In Fig. 2, the PAS signal strength does not necessarily correlate with decreases in sample loading or increases in detection distance. Part of this variability in the PAS signal strength occurs because of the PAS signal dependence on laser power. 7 For the distance studies, our PAS signals were not normalized for laser power which ranged from 60–80 mW. In addition, small variations in the collection angle of the parabolic dish impacts the PAS signal. The parabolic dish (Fig. 1b) was aligned visually. Its orientation was not further optimized. Furthermore, due to our use of a relatively small laser spot size (∼1 mm diameter) inhomogeneities in sample loading impact the PAS signal. Even with these PAS strength variabilities, we can detect all three of the explosives studied, which have different absorption band maxima around our laser excitation wavelength of 213 nm.
Influence of Photochemistry on PAS Signal Stability
The photochemistry of PETN, TNT, and AN when irradiated with deep UV light were previously studied using resonance Raman spectroscopy.21–23 With 229 nm excitation, PETN in solution undergoes cleavage of the O–NO2 bonds to form NO2 dissolved in solution. 22 In the solid state, PETN is expected to produce gaseous NO2. Bykov et al. observed that AN in the solid state continuously loses mass with 213 nm excitation without forming any new Raman bands. 23 They concluded that gaseous photoproducts were being formed. TNT, on the other hand, produces solid photodegradation products upon 229 nm excitation such as 2-amino-4,6-dinitrotolune (DNT), 4-amino-2,6-DNT, 3,5-dinitroaniline (DNA), and possibly carbonaceous species.21,25 It is important to understand the impact of this type of photochemistry on detecting trace explosives with deep UV PAS.
To determine the impact of photochemistry on the photoacoustic signal, we examined the PAS signal over 14 s time frames for AN, PETN, and TNT at a standoff distance of 2 m, as shown in Fig. 4. We see a gradual increase in signal strength for TNT, which coincides with TNT turning yellow/brown due to photodegradation (Fig. 4a). A few of the photoproducts of TNT (e.g., 4-amino-4,6-DNT and 3,5-DNA)
21
have stronger absorptions at 213 nm than TNT likely leading to the stronger PAS response.10,21,25 Therefore, longer irradiation periods will produce stronger PAS signals as TNT's photoproducts form.
Time dependence of the deep UV PAS signal for (a) TNT, (b) PETN, and (c) AN at 250 (red circle), 100 (blue diamond), and 10 (green square) µg/cm2 ink-jet printed samples on Al substrates. The signal was collected every 1 s for 14 s at a 2 m standoff distance. A gradual decrease in PAS signal with time is observed for PETN and AN; whereas, the TNT PAS signal gradually increases.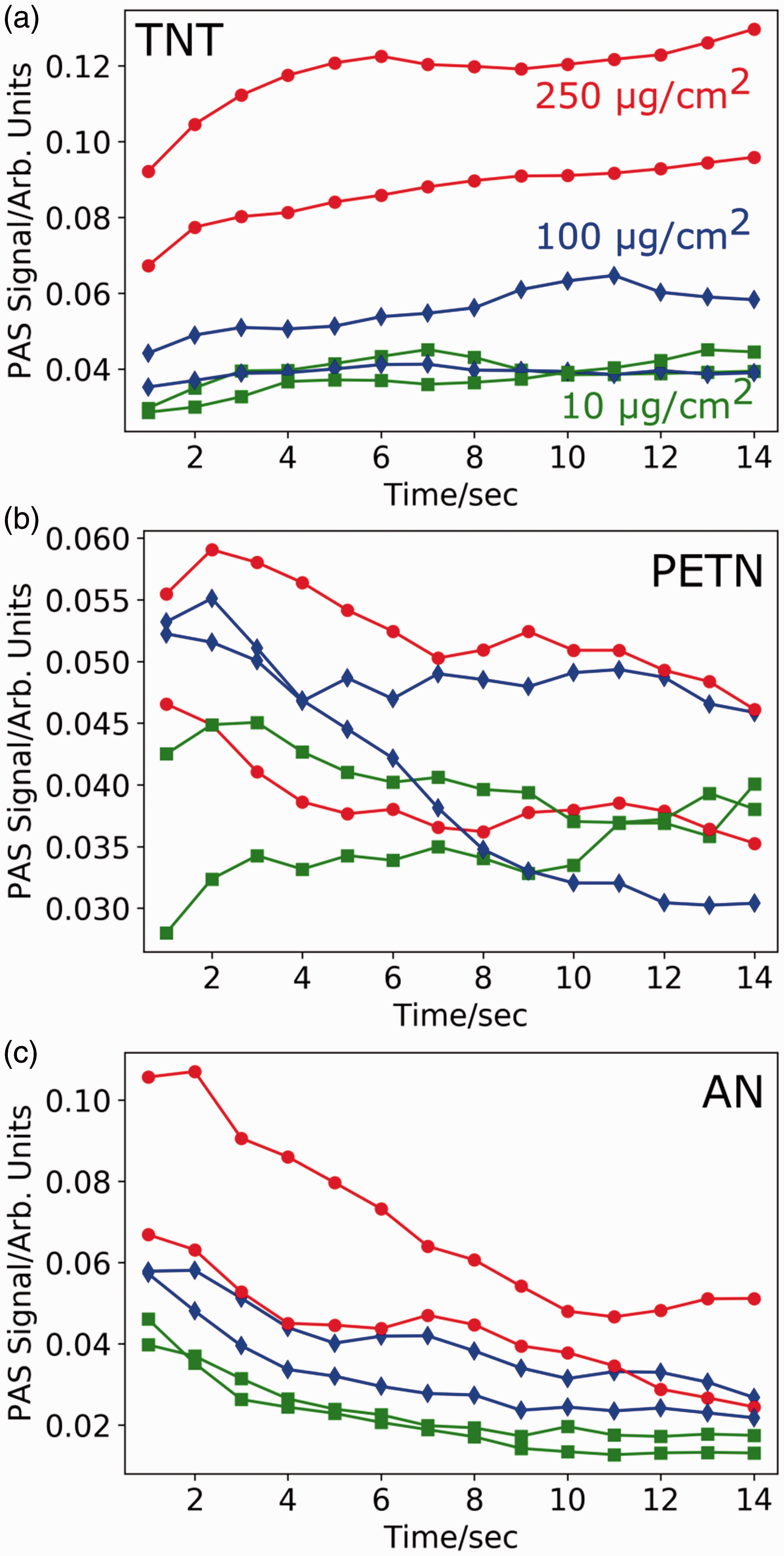
In contrast to TNT, the PAS signal decays for both AN and PETN (Figs. 4b and 4c). This coincides with a loss of mass for these analytes, due to the photochemical production of gaseous species that leave the substrate surfaces.22,23,26 At higher loadings, our PAS signal remains above the background for >14 s even as the analyte loading decreases due to photolysis (Fig. 5a). As the initial loading of the AN and PETN is decreased, the PAS signal decays faster. For example, Figs. 5b and 5c show 30 PAS acquisitions collected at a 3 m standoff for AN and PETN at 10 µg/cm2 compared to the background signal. The onset of PAS irradiation (indicated by the red line) results in a sharp increase in the PAS signal that lasts for ∼5 s before decaying into the background. At trace coverages, these explosive samples become depleted quickly, lowering the irradiation time available for observing the PAS response.
(a) Deep UV PAS signal of 250 µg/cm2 of AN and PETN on Al substrates over 14 s at 1 m. (b) PAS signal of 10 µg/cm2 of PETN over 30 s at 3 m. (c) PAS signal of 10 µg/cm2 of AN over 30 s at 3 m. For the 10 µg/cm2 samples, data collection was started before irradiating the sample. The red line indicates when the laser was unblocked. A sharp increase in PAS signal is observed over several acquisitions before the signal decays into the background.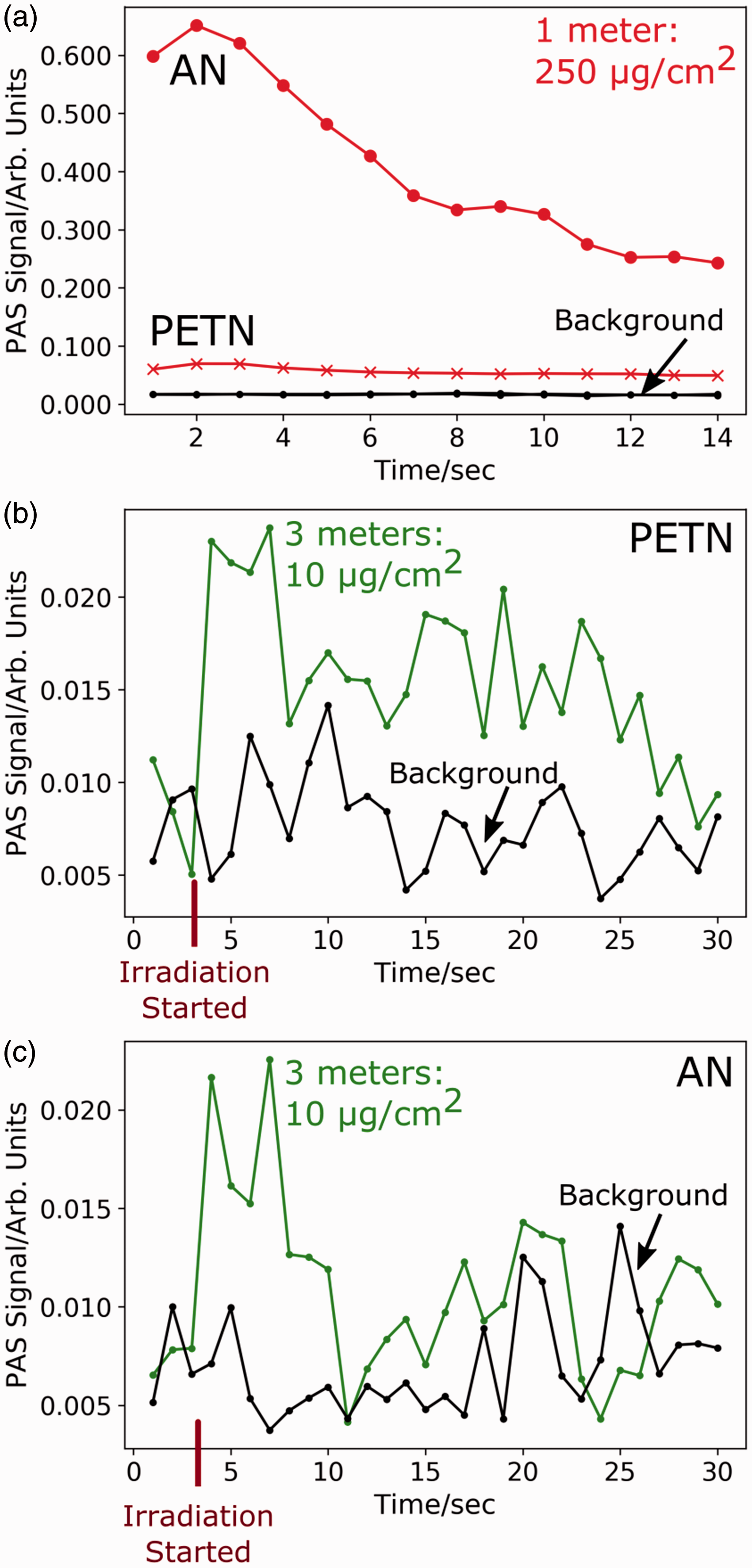
Influence of Photochemistry on PAS Signal Strength
Photochemistry has been shown by Chen et al. to provide a 3000-fold increase in PAS signal strength.
27
They compared the PAS strength of a carbon suspension that underwent high-temperature chemical reactions with that of a simple dye solution with comparable absorbance. They proposed the signal enhancement resulted from gas production. To investigate this possibility, we compared the PAS signal of AN which photochemically produces gaseous species to the nitrate salts NaNO3, KNO3, and RbNO3, as shown in Fig. 6a and Fig. S3.
(a) Deep UV PAS laser irradiance dependence of AN, NaNO3, KNO3, and RbNO3. Average and standard deviation of the PAS signal is shown at each irradiance. Ammonium nitrate has the strongest PAS signal at higher laser irradiances. (b) Laser-induced temperature versus laser irradiance of the nitrate salts. Ammonium nitrate reached the highest laser-induced temperature at ∼55 ℃ (18 W/cm2).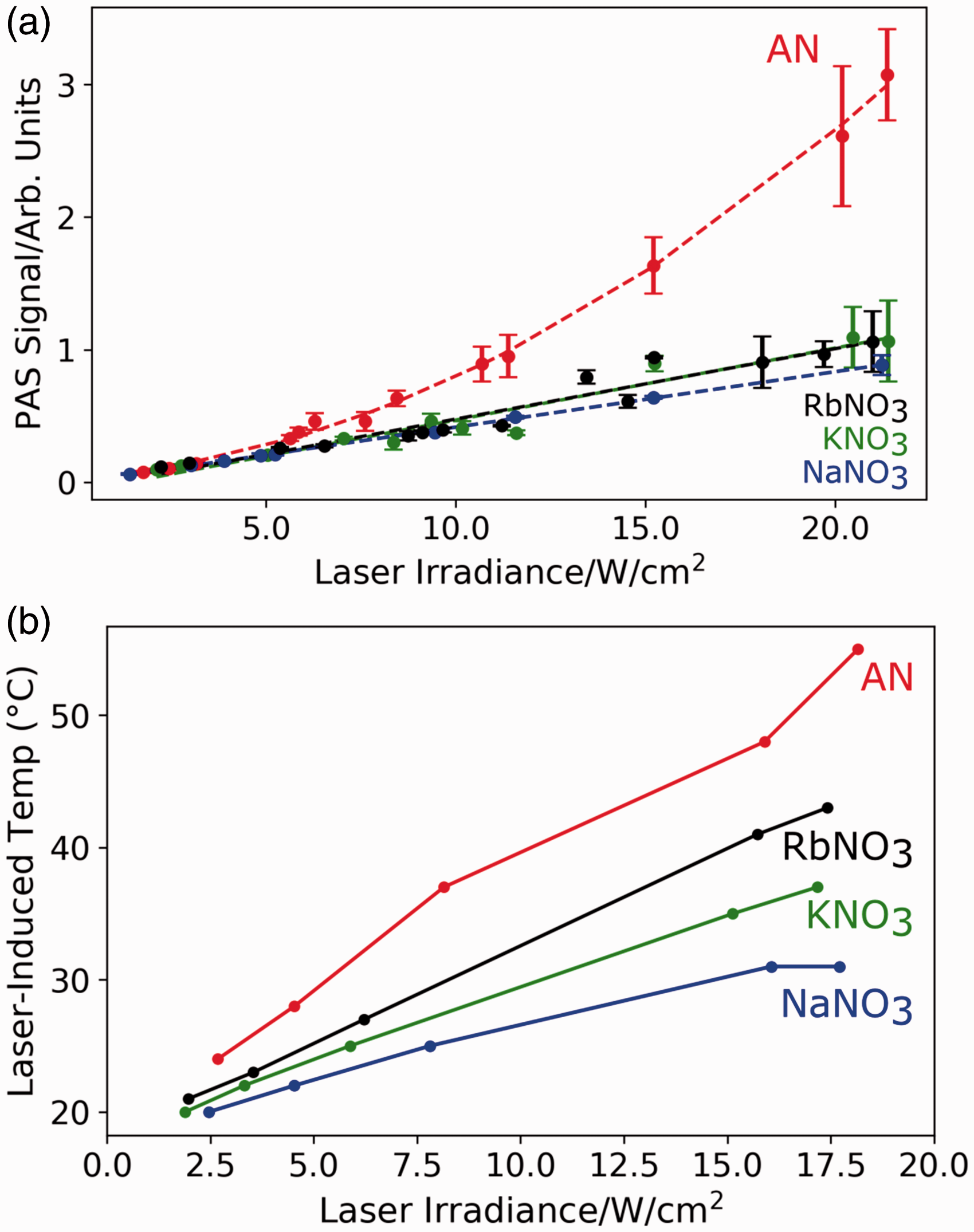
We expect the deep UV absorptions derive from identical π–π* electronic transitions of the nitrate groups. We also expect very low photolysis quantum yields for these solid-state alkali metal nitrates. For example, solid NaNO3 forms NaNO2 and O· with 299 nm irradiation with a photolysis quantum yield of ∼10−8.23,24 As this photolysis has a negligible quantum yield, we believe it will not contribute to the PAS signal. Investigations of the PAS signal stability of the nitrate salts are provided in Fig. S3.
The PAS signal of these nitrate salts are also expected to depend on their heat capacities which are 120.7 J/mol K (41℃) for AN, 98.9 J/mol K (59.7℃) for NaNO3, 99.7 J/mol K (59.7℃) for KNO3, and 108.1 J/mol K (60.7℃) for RbNO3.29–32 Given the similar nitrate absorbances, the higher heat capacity for AN predicts a weaker PAS signal. We were unable to find values for the thermal conductivities of all the nitrate salts.
Figure 6a shows that, at low laser irradiances (<5 W/cm2), the PAS signals for the nitrate salts are nearly identical. However, as the laser irradiance increases above 5 W/cm2, the AN PAS signal rapidly increases. Despite the same nitrate group absorbance and higher heat capacity, AN has the highest PAS signal by a factor of two to three at laser power irradiances >17 W/cm2. The other nitrate salts have nearly identical PAS strengths.
Above ∼10 W/cm2 AN shows a superlinear response, which presumably results from photochemical gas formation. A similar trend was observed by Wynn et al., 18 where at higher laser fluences they saw deviations from a linear PAS response that correlated with an increase in ablated material. This higher signal for AN indicates that production of gaseous species upon irradiation enhances the PAS response.
Influence of Laser Heating on PAS Strength
We also investigated the effect of laser heating on the PAS signal strength of the nitrate salts. Figure 6b shows the laser-induced temperature for AN, NaNO3, KNO3, and RbNO3 across a range of irradiances. Ammonium nitrate reached the highest laser-induced temperature of all the nitrate salts at ∼55 ℃ for a laser irradiance of 18 W/cm2. This temperature is below the AN thermal degradation temperature of ∼170 ℃. 33
To understand the impact of this temperature increase on the AN PAS signal strength, we examined the dependence of the PAS signal strength on the sample temperature for AN and NaNO3 (Fig. 7a). We heated the entire sample using thermal heating tape while recording the sample temperature using an IR camera. We used a low laser irradiance (<5 W/cm2) for this study to minimize the laser-induced temperature increase. At sample temperatures above 60 ℃, we see a decrease in the PAS signal strength for AN. This PAS signal strength decrease does not result from extended irradiation of a particular sample spot and is reversible (at least once) as shown in Fig. S4.
(a) Deep UV PAS signal dependence on sample temperature from 20 ℃ to 108 ℃. A decrease in signal is observed for AN at higher sample temperatures; whereas, NaNO3 shows little change across the range of temperatures. The PAS signal is normalized for laser irradiance. (b) The PAS laser irradiance dependence of two AN samples at ∼20 ℃ and ∼100 ℃. These are the initial temperatures of the AN samples and do not include temperature increases due to laser heating.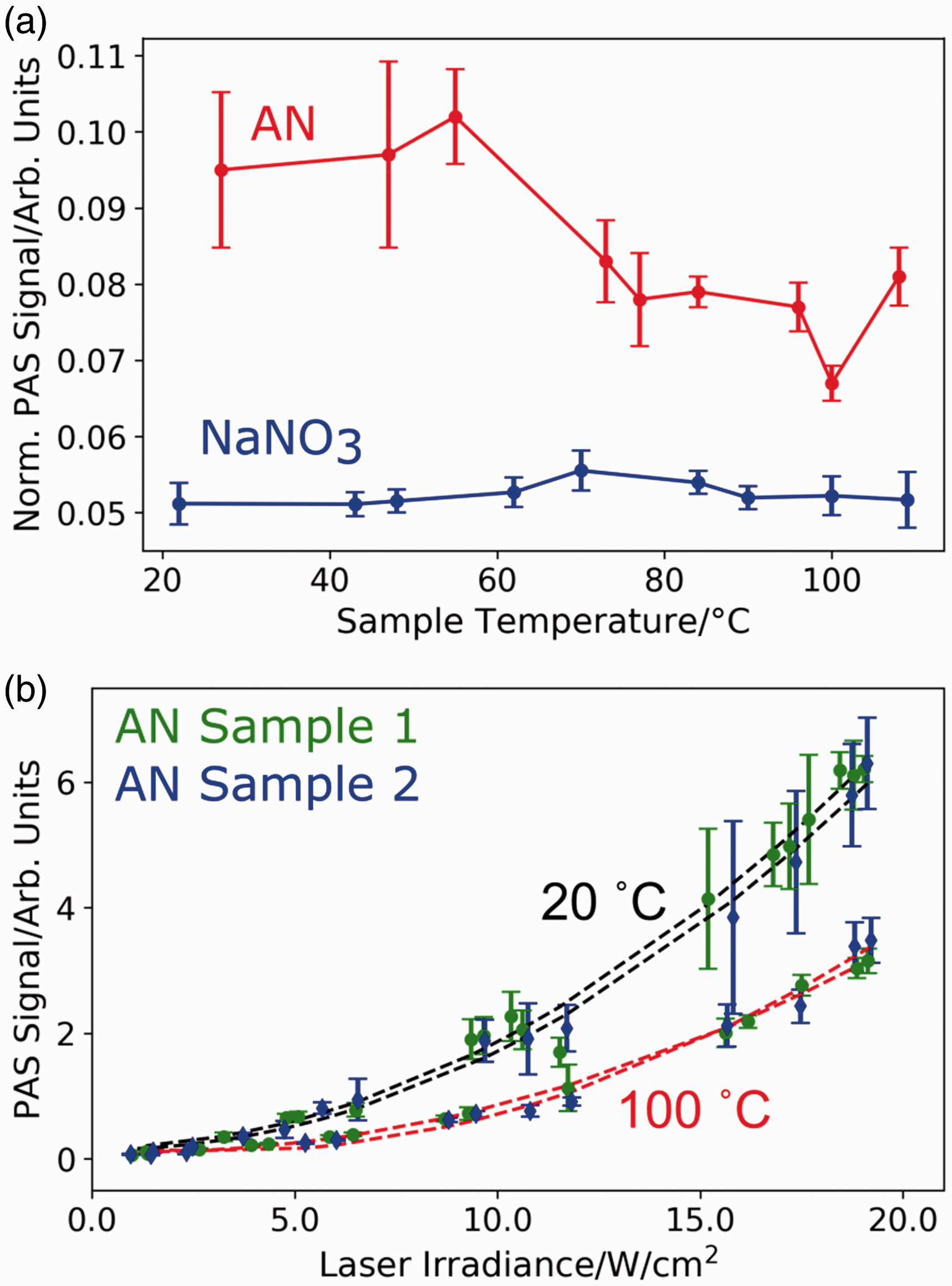
We also examined if this decrease in PAS signal strength for AN occurs over a range of laser irradiances. Figure 7b shows the PAS signal irradiance dependence of two samples at temperatures of ∼20 ℃ and ∼100 ℃. As the laser irradiance increases the PAS signal increases superlinearly for both temperature regimes. At the elevated temperature, however, the signal is weaker across the entire range of laser irradiances.
This decrease in PAS signal for AN at higher temperatures probably results from a temperature-dependent phase change of the AN crystal structure. The relevant phase transitions for AN are phases IV to III at 32 ℃, phases III to II at 84.5 ℃, and/or a metastable phase transition from IV to II at 50 ℃.32,34,35
We are unable to decide whether the III to II or IV to II phase transition is responsible for the PAS signal decrease but both phase changes result in a roughly 10% increase in heat capacity for AN leading to an expected weaker PAS signal.32,34–36 In contrast, we see no temperature dependent change in the PAS signal of NaNO3 because it does not undergo a phase change over this temperature range. 37 The decrease in signal for AN due to a change in heat capacity highlights the importance of the traditional PAS mechanism for explosive detection.
Considerations for Deep UV PAS
While we demonstrated that deep UV PAS can indicate the presence of explosives at meter standoff distances, deep UV PAS cannot currently identify the explosive detected. Development of wavelength tunable deep UV lasers would enable PAS to determine the analyte absorption spectrum providing a potential avenue for increasing the specificity.
Alternatively, deep UV PAS can be used as a prescreen technique to quickly locate explosive contamination. Once the explosive was located, a complementary technique, such as deep UV resonance Raman spectroscopy that requires longer measurement times could be used to identify the explosives. 5
Conclusion
We demonstrate deep UV PAS for the standoff detection of trace explosives, achieving 10 µg/cm2 detection of AN, TNT, and PETN at 3 m by exciting into the absorption bands of explosives with a 213 nm laser. We utilize a sensitive microphone and modest laser powers. Many explosives also undergo photochemistry when irradiated with this deep UV light. We observe that the photochemical production of gaseous species of AN enhances the PAS signal strength; however, with continued irradiation AN can become completely photolyzed, resulting in a loss of PAS signal. The fast response times of deep UV PAS enable detection of trace explosives before complete photolysis.
Supplemental Material
Supplemental material for Deep Ultraviolet Standoff Photoacoustic Spectroscopy of Trace Explosives
Supplemental material for Deep Ultraviolet Standoff Photoacoustic Spectroscopy of Trace Explosives by Alyssa B. Zrimsek, Sergei V. Bykov and Sanford A. Asher in Applied Spectroscopy
Footnotes
Acknowledgments
We thank the Office of Naval Research for financial support. We would also like to thank the Army Research Laboratory (ARL) for donating ink-jet printed explosives samples for testing our instrumentation.
Conflicts of Interest
The authors report there are no conflicts of interest.
Funding
This work was supported by the Office of Naval Research (ONR; grant number N00014-16-1-2681).
Supplemental Material
All supplemental material mentioned in the text is available in the online version of the journal.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.