
Abstract
Biomaterials are a key element of bone tissue engineering, where they can act as scaffolds for tissue regeneration. The goal of presented research is to evaluate the influence of oleic acid (OA) on the structure and properties of poly(ɛ-caprolactone), or PCL, (with molar masses: 45 000 and 80 000 g/mol), hydroxyapatite (HAp) composites and to determine the optimal OA to HAp ratio ensuring optimal material properties. Visual assessment as well as light and scanning electron microscopy revealed improved dispersion of HAp in the PCL matrix due to its amphiphilic character and, therefore, stabilisation of HAp by OA. However, excessive OA caused deterioration of the PCL45k membrane as cracks were observed. Moreover, the mechanical performance also decreased when the OA to HAp ratio was greater than 1:3 (by weight). Raman spectra confirmed that such effects were due to the increasing amorphous nature of the membranes with increasing OA concentration. Two-dimensional correlation spectroscopy (2D-COS) revealed clear differences in the response of the PCL–HAp system to increasing OA content depending on the molar mass of PCL. Considering the obtained data, the OA:HAp ratio in the PCL matrix ensuring a homogeneous structure and the highest mechanical performance is 1:6 by weight.
This is a visual representation of the abstract.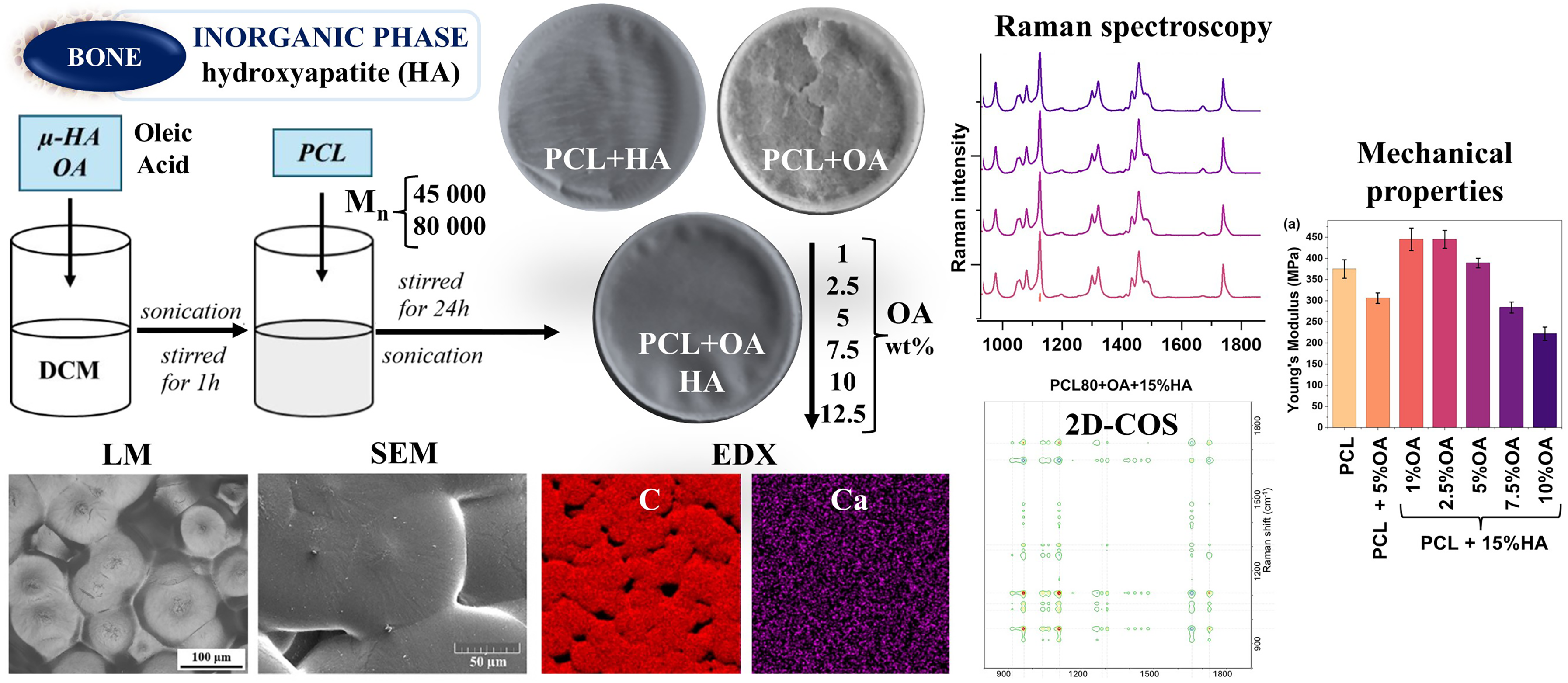
Keywords
Introduction
Skeletal injuries, including bone defects and fractures that exceed the native regenerative ability of bone tissue, are the main focus of investigation in the field of regenerative medicine. It is an interdisciplinary field that aims to repair, modify, and regenerate damaged tissues and organs. One of its main areas is bone tissue engineering, which uses biomaterials to promote tissue regeneration.1,2 These biomaterials, usually in the form of scaffolds, have to be biocompatible, exhibit optimal mechanical performance, and have appropriate structural characteristics (such as porosity) to support native cell migration and proliferation, ultimately leading to regeneration of damaged tissue.3,4 These types of biomaterials should also exhibit biodegradability in a physiological environment and be non-toxic to avoid triggering an immune response in the body. 5 Due to the diverse requirements imposed on scaffolds, composite materials, consisting of two or more constituents that individually exhibit desirable properties, but also possess inherent limitations, are widely employed in bone tissue engineering. By combining these components, it is often possible to overcome the shortcomings of each individual material while synergistically enhancing their overall performance. 1 Although many biomaterials have been studied in recent years, only a few of them become part of therapeutic strategies in practical medicine.6–8
Hydroxyapatite
One of the main components of biomaterials used in bone tissue engineering is hydroxyapatite (HAp), with the molecular formula: Ca10(PO4)6(OH)2. HAp constitutes the inorganic part of bone extracellular matrix, and its chemical composition enables the release of calcium and phosphate ions, which can be subsequently absorbed and metabolized by the organism. 9 Its ion-exchange capacity, as well as the possibility of surface functionalization, allow the design of scaffolds capable of exchanging ionic and molecular components. 10 This behaviour is associated with key properties of HAp, which are osteoconductivity and bioactivity, which stimulate the growth and continuity of regenerated and native tissues. Hydroxyapatite, in addition to being biocompatible, is characterized by low water solubility and high physicochemical stability, which makes it a very promising material for a wide range of biomedical applications.9,11
Poly(ɛ-caprolactone)
Poly(ɛ-caprolactone) (PCL) is an aliphatic polymer obtained via ring-opening polymerisation of cyclic ɛ-caprolactone. PCL has been approved by the U.S. Food and Drug Administration (FDA) and, due to its compatibility and non-toxic profile, it is already widely employed in medical applications such as resorbable sutures and as a matrix for tissue scaffolds. 12 PCL is biodegradable, as it hydrolyse to 6-hydroxycaproic acid, and its complete degradation in a biological environment generally takes 2–3 years, depending on factors such as molecular weight, structure, and residual monomer content. 13 The degradation rate of materials used to manufacture scaffolds in tissue engineering is particularly important because if it is too slow, it hinders new tissue formation; if it is too fast, it can result in premature loss of mechanical integrity and disrupt integration between scaffold and tissue. 14 However, PCL has certain limitations associated with its relatively low thermal stability (melting point 59–64 °C), which reduces its long-term durability. 15 In addition, the hydrophobic properties of PCL diminish cell adhesion and proliferation, resulting in low biocompatibility, and thus limit its broader biomedical applications. Therefore, further modifications of the PCL matrix are crucial to enhance its biocompatibility, reduce hydrophobicity, and improve mechanical performance.
Oleic Acid
In order to achieve proper integrity and uniformity of composites, additive substances are employed to promote the dispersion of components throughout the polymeric matrix. Oleic acid (OA) is a monounsaturated fatty acid that is a colourless and odourless substance under standard conditions. Its molecule consists of a hydrophobic moiety in the form of a long aliphatic chain and a hydrophilic moiety, which is a carboxyl group. Due to such amphiphilic architecture and the resulting surface-active properties, OA can act as a surfactant, enhancing the interaction between hydrophobic and hydrophilic phases in composite. 16 Therefore, in the PCL–HAp composites, the addition of OA is expected to improve the dispersion of the HAp, the inorganic phase (OA is suspected to bind to the surface of HAp powder through chemisorption and form oleate complexes) in the hydrophobic PCL polymer matrix, the organic phase. Stabilization of the HAp surface by OA and the expected increase in the homogenous HAp dispersion, should lead to higher bioactivity, cohesion and mechanical performance of the resulting composites. From a biochemical point of view, OA plays an important role in cellular metabolism. 17 According to medical research, diet rich in oleic acid reduce cardiovascular risks, increase high-density lipoprotein (HDL) and decrease low-density lipoprotein (LDL) levels. 18 Oleic acid also helps reduce the level of inflammatory cell adhesion molecules, leading to reduced inflammation. 19 This property of OA is promising in the context of modifying composites used in tissue engineering, as the reduction of the expression of adhesive molecules limits the inflammatory response after implantation, thus improving the biocompatibility and integration of the scaffold.
Research Objectives
To our knowledge, studies on PCL composites containing selected concentrations of OA have primarily addressed their antioxidant and antibacterial properties, as well as the influence of OA on viscoelastic behaviour and on the electric current and viscosity relevant to melt electrospinning, with relatively limited attention devoted to structural or mechanical characteristics.20–22 So far, we found only three publications that investigated PCL/HAp/OA composites in details, namely those by Kim et al., 23 Cardoso et al., 24 and Lin et al. 25 The authors demonstrated that the presence of OA promotes a more homogeneous dispersion of HAp in the PCL matrix, which leads to improved mechanical and biological properties. It is noteworthy that their investigation was limited to composites prepared using only one OA:HAp ratio (such as 1:12 or 1:3.5).23,25 Taken together, these studies clearly indicate that PCL/HAp/OA composites represent a promising class of scaffold materials for bone regeneration, due to their increased mechanical strength, better micro/nanoparticle dispersion, and favourable cellular interactions. Nevertheless, no investigations have systematically addressed to optimize the HAp to OA ratio, especially as a function of the PCL molar mass, which is a key parameter to achieve a high degree of material homogeneity and to tailor the structural and mechanical properties of the composites.
In this study, two series of composite materials consisting of a polymer matrix of PCL, HAp, and OA were fabricated. The two series of materials prepared differed in the molar mass of PCL: PCL (Mn = 45 000) + OA + HAp and PCL (Mn = 80 000) + OA + HAp. In both series, the HAp content was set at 15 wt%, while the OA content was varied and set to 1, 2.5, 5, 7.5, 10, and 12.5 wt%, respectively.
The novelty of this work and the main aim of the research was to systematically analyse of the influence of the presence and increasing concentration of OA on the dispersion of HAp in polymer membranes, as well as on the resulting mechanical and physicochemical properties of the obtained polymer composites. Membranes were produced by the solvent casting method, and their morphology was examined by polarized light microscopy (PLM) and scanning electron microscopy (SEM). Additionally, scanning electron microscopy–energy dispersive X-ray spectroscopy (SEM-EDX) elemental mapping (C, O, P, Ca) was performed to assess the spatial distribution of the inorganic phase. Determination of Young’s modulus and tensile strength enabled evaluation of the effect of increasing OA concentration, in relation to the constant HAp content, on the mechanical properties of the materials. Raman spectroscopy was selected as the primary technique to assess the impact of OA concentration on the degree of crystallinity of the polymer matrix. Therefore, the spectroscopic crystallinity parameters were quantified based on the curve fitting (deconvolution) of amorphous and crystalline contributions within the same vibrational bands. In order to obtain more detailed insight into the interactions between OA, HAp, and the PCL matrix, especially as a function of PCL molar mass, two-dimensional correlation spectroscopy (2D-COS) was employed, treating the OA concentration as an external perturbation. At the same time, the research presented in this paper demonstrates the usefulness and effectiveness of 2D-COS analysis in the field of materials science.
Experimental
Materials and Methods
Used for this work were PCL (Mn 45 000 g/mol and Mn 80 000 g/mol, Sigma-Aldrich); HAp (Ca5(OH)(PO4)3, particle size <2.5 µm ± 0.5 µm (Sigma-Aldrich); OA (ThermoScientific); dichloroethane (DCM; POCH S.A.).
Sample Preparation
The polymer membranes were fabricated using a solvent casting method. HAp (15 wt% of final material) was added to OA (1, 2.5, 5, 7.5, 10, and 12.5 wt% of final material), solution in 4 cm3 DCM. The obtained mixture was subjected to multi-stage mixing: sonication with ultra-sonic homogenizer (3 minutes, amplitude 30%; Sonics and Materials, Inc., VCX130) and mixing on magnetic stirrer for 1-h. Then, PCL granules were added to the mixture and left to mix on magnetic stirrer for 24-h at room temperatures. In the final stage, the prepared suspensions were sonicated again (3 min, 30% amplitude, Sonics and Materials, Inc., VCX130) and immediately poured into Petri dishes (6.5 cm in diameter) using a pipette. The Petri dishes were covered with Parafilm with holes to prevent excessive solvent evaporation and left for at least 24-h at room temperature for polymer to crystallize. For each polymer molar mass, Mn 45 000 g/mol and Mn 80 000 g/mol, a separate series of composite membranes was prepared. Composites without oleic acid, made only of polymer (PCL) were used as reference materials. The diagram of the prepared samples is presented in Figure 1.
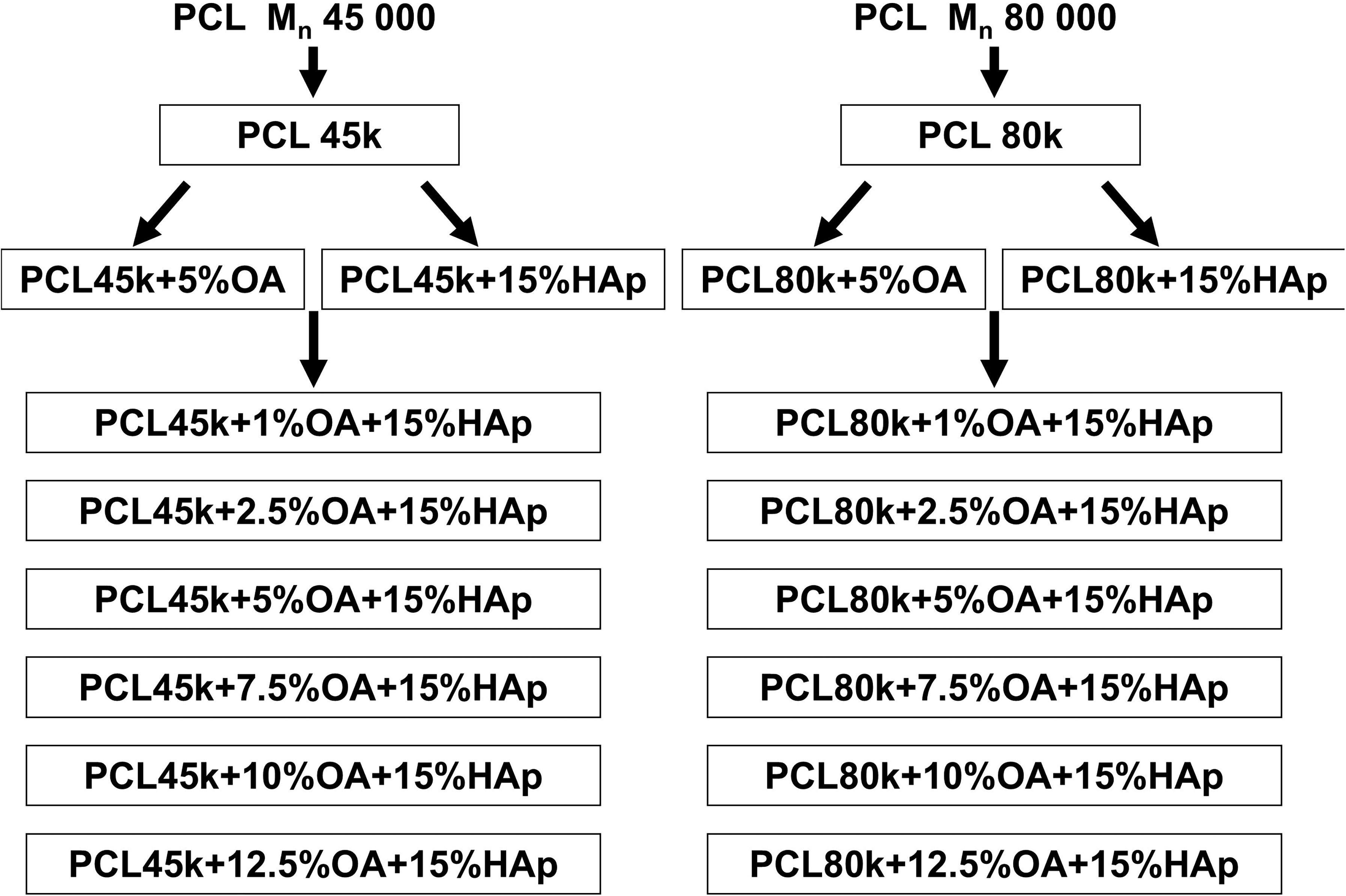
Diagram of prepared composite membranes.
Polarized Light Microscopy
Polarized Light microscopy (PLM) micrographs were acquired using a Nikon Eclipse 80i microscope. Thin sections of the samples (thickness 5 μm) were prepared with a rotary microtome RM 2255 (Leica, Vienna, Austria), placed in a thin layer of immersion oil between glass slides, and examined under transmitted light.
Scanning Electron Microscopy (SEM)
Scanning Electron Microscopy images were obtained using a microscope MAIA3 (Tescan, Czech Republic) equipped with an X-MaxN 20 detector for EDX (Oxford Instruments, UK). All specimens were fixed on a metallic holder using conductive carbon tape, then sputter-coated with a thin platinum film (vacuum sputter coater SCD 050; Leica, Austria) and examined by secondary electron imaging (SEM/SE) at an accelerating voltage of 3 kV. The EDX spectra (elemental microanalysis) were acquired at a higher accelerating voltage of 30 kV.
Mechanical Tests
Tensile tests were performed using an Instron 6025/5800R universal testing machine (Instron Ltd., UK) with a cell load of 100 N and an applied speed of 10 mm/min (at room temperature). Dumbell-shaped specimens of ISO527-3/5 type, half-size (total length 60 mm and width of narrowed part 3 mm; thickness 0.1 mm) were tested. For each sample, the results of four individual measurements were averaged.
Raman Spectroscopy
Raman spectroscopy spectra were taken using the Renishaw inVia Qontor Raman spectrometer (UK) coupled to a Leica DM2700 optical microscope. The measurements were performed using an HPNIR laser with a wavelength of 785 nm with its power adjusted so that the material is not disturbed or destroyed. The laser beam was focused at 50× magnification using a long-range objective. Raman radiation was analysed using a diffraction grating with 1200 slits per 1 mm. Spectra were collected in the range from 2000 cm−1 to 200 cm−1, with an exposure time of 10 s and four accumulations for each spectrum. To improve the signal-to-noise ratio, spectra from five measurements spots for each sample were averaged followed by a cosmic ray removal, smoothing, baseline correction, and curve fitting (Renishaw, WiRE v.3.4 and v.5.5 and Thermo Scientific, OMNIC v. 7.3 software).
Three spectral ranges were selected in which curve fitting was performed: (i) Range from 1340 cm−1 to 1250 cm−1 (two-band model), I1285/I1306 ratio (Figure S1a, Supplemental Material), (ii) range from 1130 cm−1 to 1020 cm−1 (four-band model), I1111/I1097 ratio (Figure S1b), and (iii) range from 990 cm−1 to 830 cm−1 (three-band model), I915/I869 ratio (Figure S1c).
Two-Dimensional Correlation Spectroscopy (2D-COS)
The two-dimensional correlation spectroscopy analysis (2D-COS) was performed according to the Noda method.26,27 The correlation maps were obtained based on the input data, which were the Raman spectra of PCL + XOA+15% HAp composites with increasing OA concentration (X = 1, 2.5, 5, 7.5, 10, and 12.5 wt%). Two separate sets of synchronous and asynchronous maps were generated, one for the PCL45k + XOA+15%HAp sample series, and second for PCL80k + XOA+15% HAp series. The increasing concentration of OA was regarded as an external perturbation. Prior to 2D-COS analysis all average Raman spectra were subjected to baseline correction using Thermo Scientific OMNIC v.7.3 software, smoothed with a Savitzky–Golay function of two polynomials and 17 points, and normalized based on the 1440 cm−1band (to remove the nonselective effect). These data preprocessing steps increase the absolute values of the cross-peaks and help to avoid erroneous determination of their signs. The correlation maps were generated by Spectra Corr v.1.1 package within Omnic software (number of contours = 12). The 2D-COS analyses were performed in the spectral region from 1900 cm−1 to 800 cm−1.
Results and Discussion
Visual Evaluation of Samples
The initial stage of sample evaluation included macroscopic visual inspection. Photographs of the two series of polymer composites (PCL45k and PCL80k series) are presented in Figure 2. The membrane made of pure PCL45k exhibits a smooth and uniform surface. The incorporation of HAp (PCL45k + 15%HAp) results in the formation of elongated aggregates. The membrane containing PCL45k and 5 wt% OA (PCL45k + %OA) displays visible cracks, indicating reduced mechanical integrity. In contrast, the composite membranes of PCL45k containing 15 wt% HAp and 1–5 wt% OA show a macroscopically homogeneous structure without visible aggregates or mechanical damage. However, at higher OA concentration (7.5–12.5 wt%) of this PCL45k series, although the cracks are not very visible in the photographs, the membranes tend to crack and crumble during handling. These observations suggest that the addition of OA improves the macroscopic homogeneity of the samples, as no HAp aggregation is observed. However, the HAp to OA ratio has to be carefully selected, as a large amount of OA leads to its free presence in the membrane, which may promote cracking and structural degradation.

Photographs of polymer membranes: (a) PCL45k series and (b) PCL80k series.
In the PCL80k series of samples, a uniform and smooth structure was observed for all membranes except PCL80k +15%HA. In this case, incorporation of HAp in the absence of OA led to the formation of spherical aggregates irregularly distributed on the membrane surface. Due to the higher molar mass of the polymer in comparison to the PCL45k series, and therefore higher density and ductility, no cracks were observed in the membranes of this series.
Morphology of Composite Membrane Surface
Polarized light microscopy (PLM) and SEM techniques were employed to evaluate the surface morphology of the composites (Figure 3). Characteristic polymeric spherulites are clearly observed. In general, spherulites consist of crystalline lamellae (folded polymer chain) that grow radially from the centre, and amorphous regions distributed between lamellae. 28 The incorporation of HAp into PCL matrix results in a significant decrease in the diameter of the spherulites, presumably because HAp acts as a nucleating agent, which causes the limitation of spherulite growth in contact with adjacent spherulites. However, adding only OA to the PCL structure results in an increase in the spherulite diameter. A similar OA effect is also seen across the entire membrane series with increasing concentration of OA, where the spherulites became progressively larger with higher OA content. In addition, SEM images show that the fibrous structure becomes more visible, especially in PCL80k series. This is presumed to be due to the increasing proportion of amorphous regions distributed between the lamellar domains. Moreover, in the case of PCL45k + 12.5%OA + 15%HAp membrane, PLM microphotograph revealed that excess OA, which is not associated with HAp, is visible along the edges of the spherulites (Figure 3a). The changes in spherulite size follow the same trend for both PCL molar mass series, PCL45k and PCL80k, respectively. However, the spherulites are smaller in the case of the PCL80k-based membrane than those observed in the corresponding PCL45k-based membrane.

(a) Polarized light microphotographs. (b) SEM images of polymer membranes.
Additionally, energy dispersive X-ray (EDX) mapping analysis was performed for elements such as carbon, oxygen, phosphorus, and calcium. The full set of results can be found in the Supplemental Material (Figure S2), whereas Figure 4 shows SEM-EDX mapping images of carbon and calcium (acquired in the same area for each sample) for the selected membranes. EDX elemental mapping of Ca and P confirmed the presence of HAp in close proximity to the membrane surfaces. Considering that the upper surface of membranes was tested, it can be concluded that HAp was not deposited during polymer crystallization. Optimally homogeneous HAp dispersion, as evidenced by distribution of Ca and P, was achieved in the case of membranes modified with 2.5 and 5 wt% OA.

Carbon and calcium elemental mapping using SEM-EDX (same location for each sample).
Mechanical Test
Young's modulus and tensile strength of the membranes made of pure PCL80k were 375 and 14.6 MPa, respectively (Figures 5a and b). Modifying PCL80k with only OA negatively affected both mechanical parameters. However, modification of PCL80k with the addition of 15%HAp and 1–2 wt% of OA results in higher Young's modulus values (up to 445 MPa) and a slight change in tensile strength (within standard deviation). After increasing the OA content to 5–10 wt%, the mechanical properties of the composites gradually deteriorated, which ultimately resulted in a Young's modulus of 222 MPa and tensile strength of 7.75 MPa for the PCL80k + 10%OA + 15%HAp membrane. Comparison of PCL+5%OA with PCL+5%OA + 15%HAp reveals beneficial influence of HAp on the membrane’s mechanical properties. Mechanical tests were carried out only on selected membranes from the PCL80k series. Samples from the PCL45k series containing 5–12.5 wt% OA showed excessive brittleness and were not tested. During handling, these samples fell apart and cracked, making their mechanical characterization difficult (as described earlier in the Visual Evaluation of Samples section above).
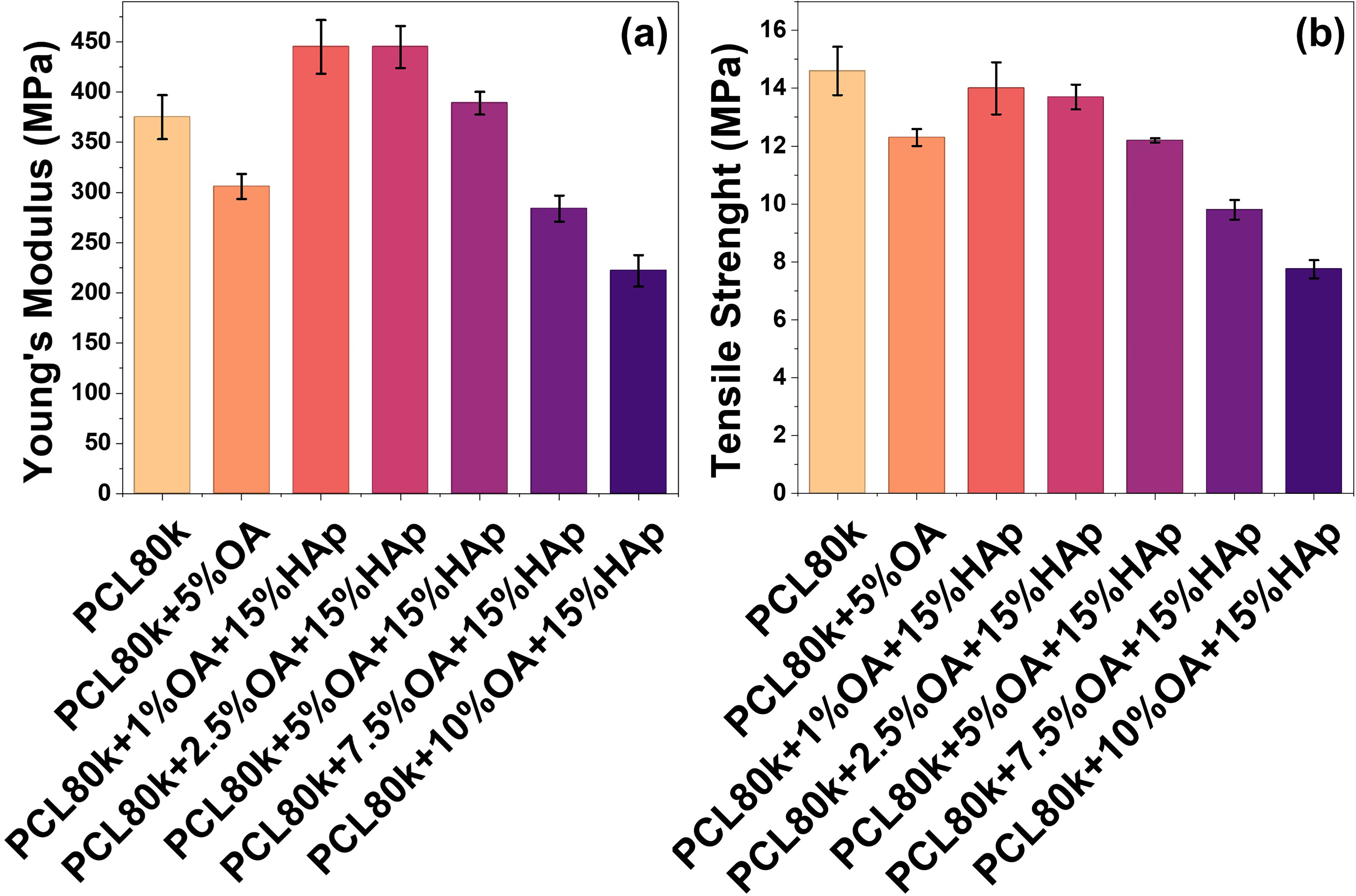
(a) Young’s modulus. (b) Tensile strength parameters of tested composites.
Raman Spectroscopy
Raman spectroscopy was used to confirm the chemical composition and determine the molecular structure of the prepared composites. Figure 6 illustrates the Raman spectra of the membranes (PCL45k series Figure 6a, PCL80k Figure 6b), as well as the OA spectrum (Figure 6c) and the HAp spectrum (Figure 6d).
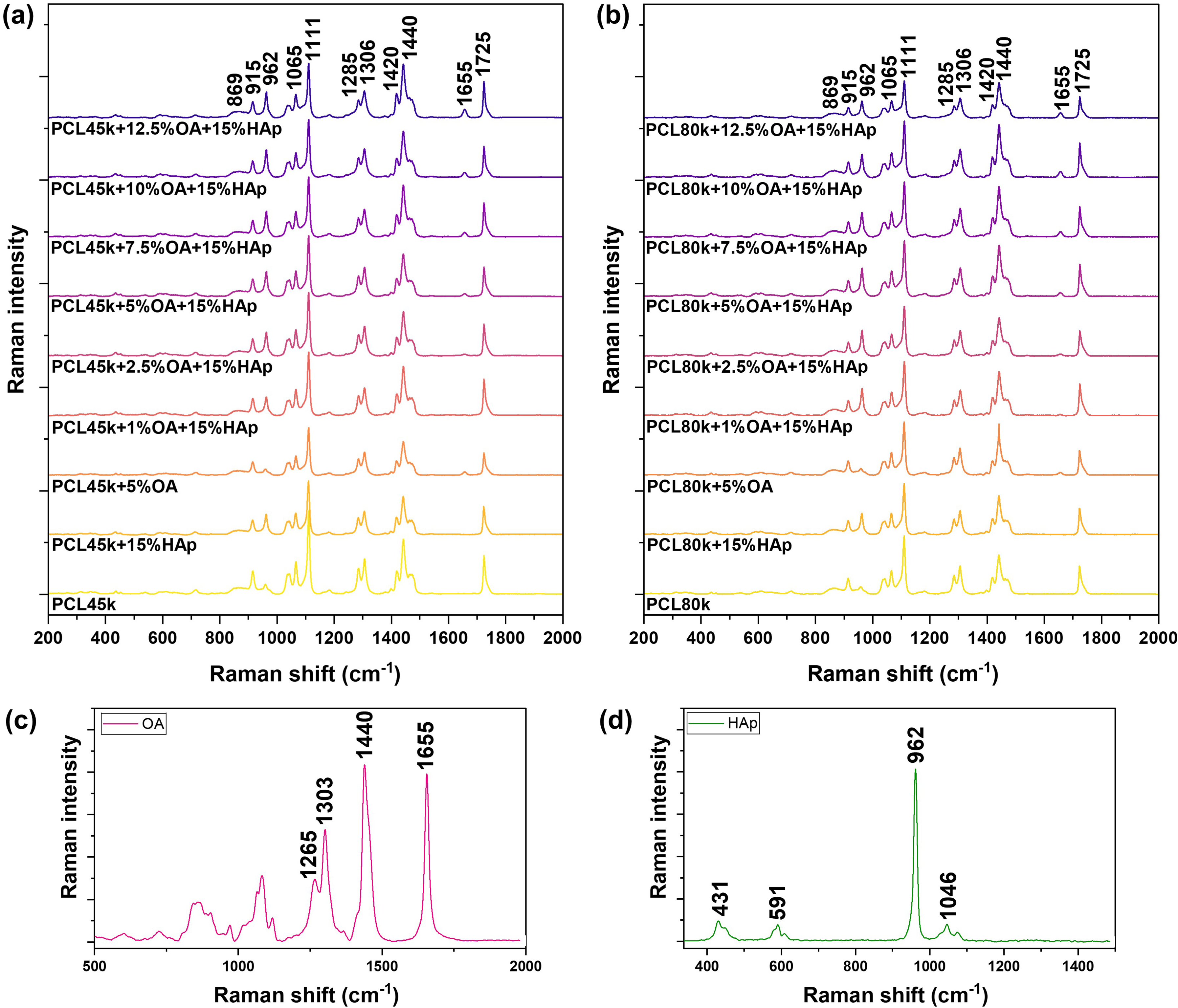
The average Raman spectra of (a) PCL45k + OA + 15%HAp composite series, (b) PCL80k + OA + 15%HAp composite series, (c) oleic acid, and (d) hydroxyapatite.
Characteristic bands for PCL are observed. The bands in the region from 1000 cm−1 to 800 cm−1 are assigned to ν(C–OOO) stretching vibrations of PCL, noting that the band at 869 cm−1 comes from amorphous polymer domains, while the band at 915 cm−1 comes from crystalline domains. The region from 1150 cm−1 to 1000 cm−1 corresponds to the ν(C–O–C) vibrations, the band at 1111 cm−1 comes from crystalline domains, while its shoulder at around 1097 cm−1 is the signal from the amorphous form of PCL. The next bands located at 1285 cm−1 (crystalline form) and 1306 cm−1 (mixture of crystalline and amorphous forms) correspond to the τ(CH2) twisting vibration of PCL. Another characteristic Raman band of PCL is located at 1725 cm−1 and is due to the ν(C = O) stretching. This band is asymmetric because it obscures the 1735 cm−1 band shoulder from the amorphous form of PCL.
To establish the assignments and spatial positions of the hidden amorphous bands, in addition to an extensive review of the literature,29–34 second derivative spectra were computed. The corresponding spectral regions, along with their enlarged views, are presented in the Supplemental Material in Figures S3–S5. Moreover, the positions of the amorphous bands were confirmed by locally melting the PCL membrane and measuring its Raman spectra, as shown in Figure S6 of the Supplemental Material. This procedure enabled an unambiguous classification of the spectral positions of the amorphous bands.
Additional bands appear in the spectra of composites derived from the used modifiers: HAp and OA. The band located at 962 cm−1 is attributed to the ν1sym(P–O) stretching vibrations of the PO43− group, which confirms the presence of HAp. Although the PCL band at a comparable wavenumber is observed in composites without HAp (ν(C–COO) band at 958 cm−1), the HAp band is easily distinguishable due to its higher intensity and slight blue shift. 29 The presence of oleic acid (OA) in the composites is confirmed by the appearance of a band at 1655 cm−1, which corresponds to the ν(C=C) cis stretching vibration of OA. As expected, the intensity of this band increases with increasing OA concentration in the composite. All Raman bands and their assignments are collected in Table I.
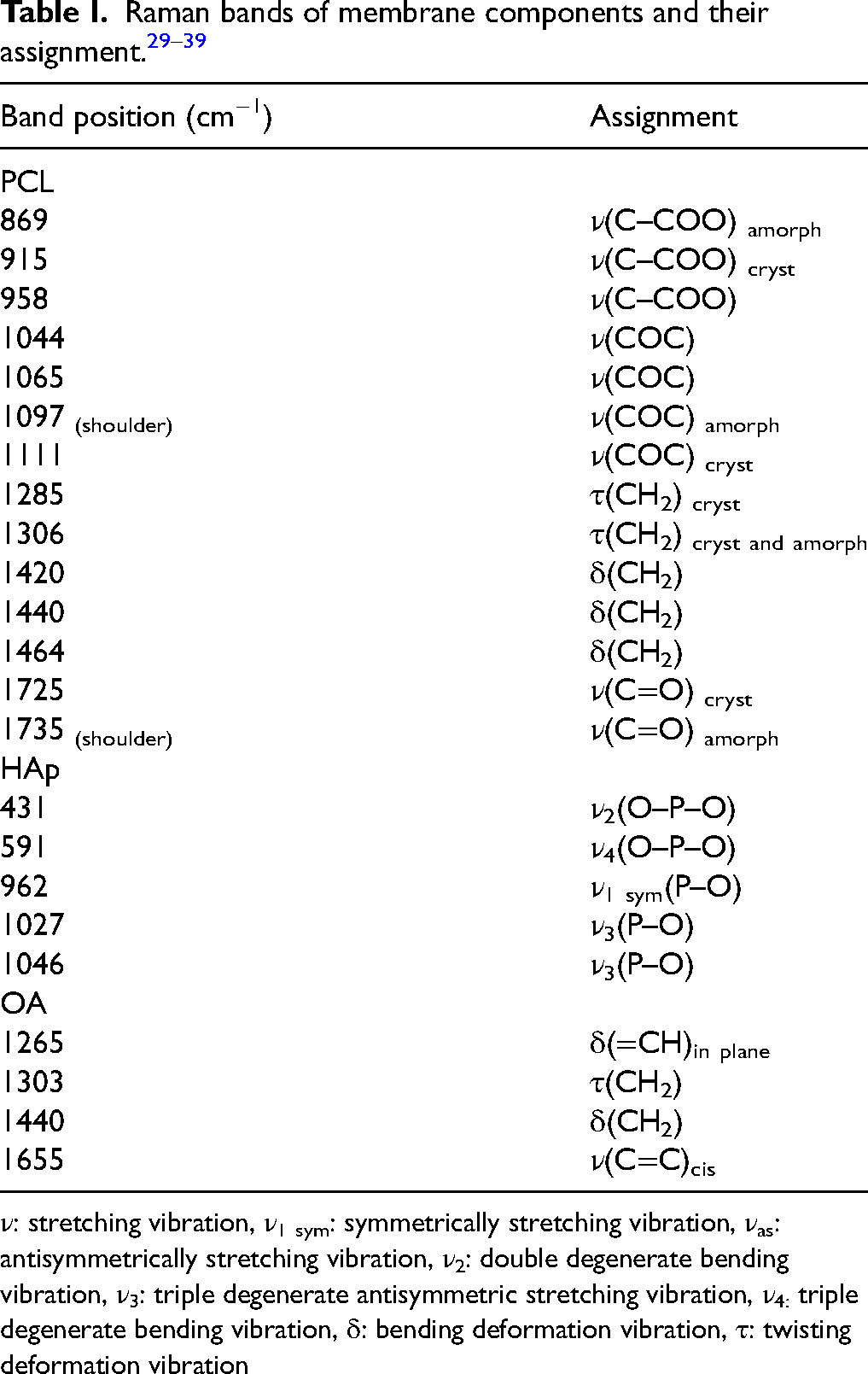
ν: stretching vibration, ν1 sym: symmetrically stretching vibration, νas: antisymmetrically stretching vibration, ν2: double degenerate bending vibration, ν3: triple degenerate antisymmetric stretching vibration, ν4: triple degenerate bending vibration, δ: bending deformation vibration, τ: twisting deformation vibration
Enlarged views of the Raman spectral regions corresponding to the amorphous and crystalline band pairs of PCL are presented in Figure 7. In each of the three chosen regions, a gradual increase in the relative intensity of the amorphous band with respect to the crystalline band with increasing OA concentration is clearly visible. To assess the crystallinity of the produced membranes, the ratios of the intensity of the characteristic bands of the crystalline domains to the bands of the amorphous PCL domains (for given type of vibration) were calculated. It was observed that increasing the concentration of oleic acid leads to the decrease in the level of polymer crystallinity, especially in the case of PCL45k series. Moreover, all band intensity ratios for PCL45k membranes are higher than the corresponding ratios for PCL80k membranes, confirming the greater amorphous nature in the case of the higher molar weight of the PCL matrix. 32
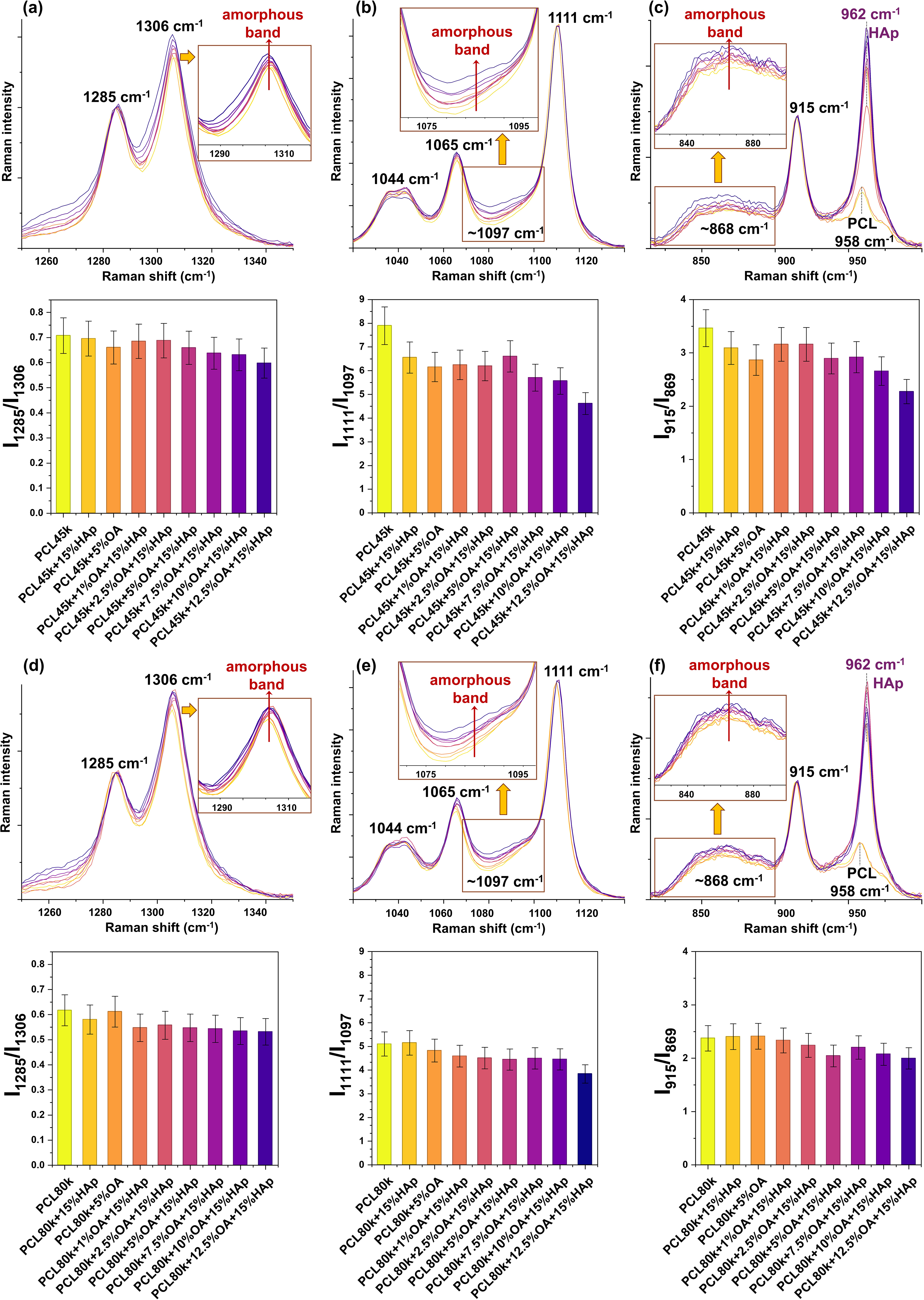
Enlarged views of the Raman spectra illustrating the amorphous-crystalline band ranges and spectroscopic polymer crystallinity parameters (ratio I crystalline /I amorphous ) for PCL45k + OA + 15%HAp series: (a) 1350 cm−1–1250 cm−1, I1285/I1306 ratio. (b) 1140 cm−1–1020 cm−1, I1111/I1097 ratio. (c) 1000 cm−1–780 cm−1, I915/I869 ratio; for PCL80k + OA + 15%Hap series: (d) 1350 cm−1–1250 cm−1, I1285/I1306 ratio. (e) 1140 cm−1–1020 cm−1, I1111/I1097 ratio. (f) 1000 cm−1–780 cm−1, I915/I869 ratio. For each region spectra were normalized to crystalline bands: (a) 1285 cm−1, (b) 1111 cm−1, and (c) 915 cm−1.
Two-Dimensional Correlation Spectroscopy (2D-COS)
To obtain additional insight into the effect of OA on the polymer structure depending on the molar mass of PCL, a two-dimensional correlation analysis was performed, treating the increasing concentration of OA as an external perturbation. The corresponding synchronous and asynchronous correlation maps are shown in Figure 8. Close-ups of narrower spectral regions can be found in Figures S7–S8 (Supplemental Material).
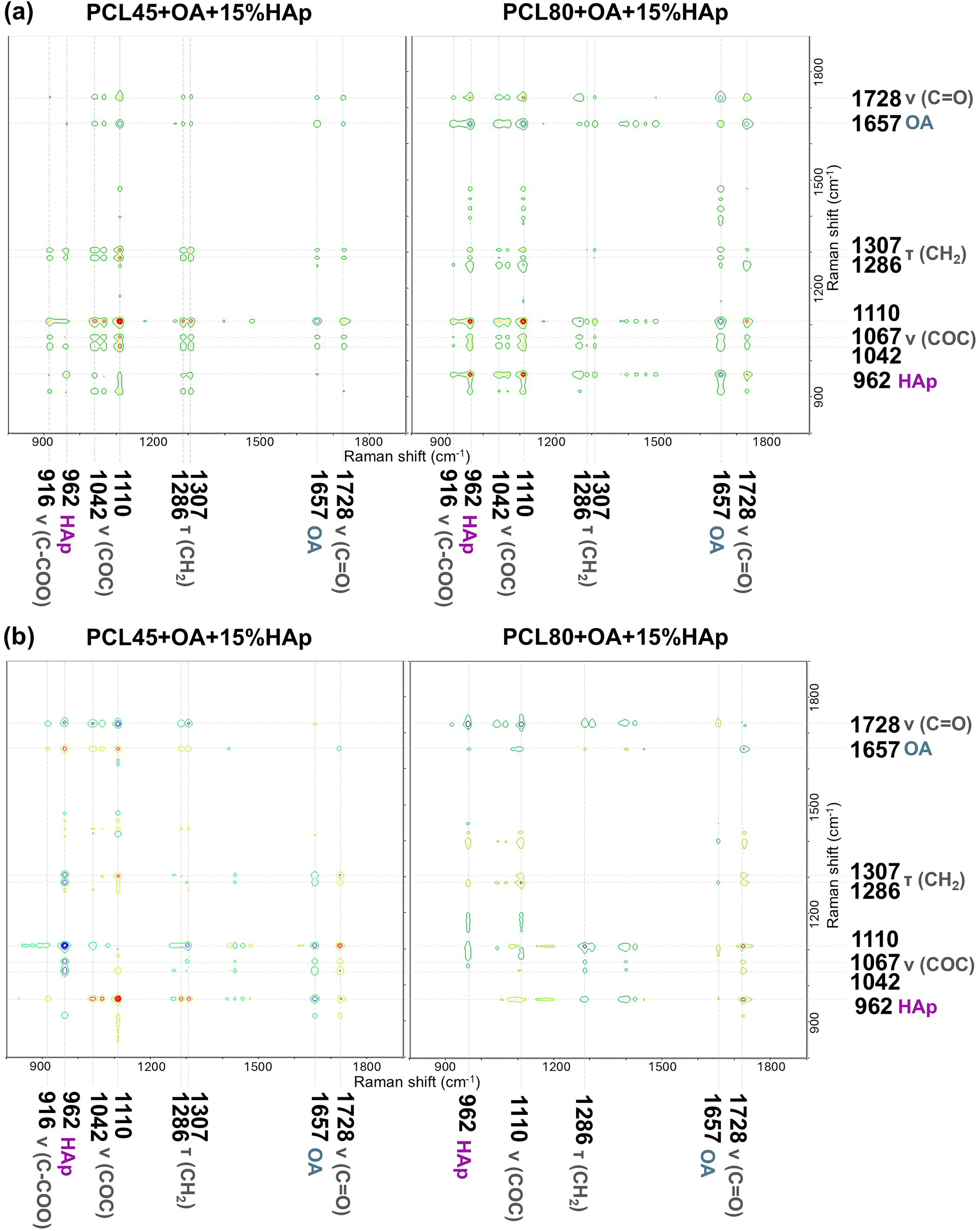
(a) Synchronous and (b) asynchronous 2D correlation Raman spectra in the wavenumber range from 1900 to 800 cm−1. Captions in grey indicate PCL vibrations, in violet HAp vibration, and in blue OA vibration. The concentration of OA in composite material was regarded as an external perturbation for molar mass of PCL composite series.
The synchronous maps for both polymer molar masses are very similar (Figure 8a). The most intense auto peak comes from the polymer band at 1110 cm−1 which is an indicator of crystallinity and reflects the structural changes occurring in the polymer after the addition of OA. The negative cross-peak arises from the simultaneous changes of the polymer band at 1110 cm−1 and the OA band at 1657 cm−1, indicating that the intensity that the changes in the OA band intensity occur in the opposite direction to changes in the polymer band. A similar interpretation applies to the remaining cross-peaks of the OA and polymer bands, which clearly demonstrates that the increasing OA concentration is the cause of the observed changes in the polymer structure. This conclusion is supported by the presence of exclusively positive cross-peaks resulting from the correlation between different bands of the PCL crystal structure, implying that all these bands change in the same direction under the influence of applied perturbation. In addition, the PCL80k + OA + 15%HAp map also shows cross-peaks that comes from the coupling of HAp and OA bands (negative) and HAp and PCL (positive), which allows for a better understanding of the coupled spectroscopic responses within the multicomponent system and suggests that increasing concentration of OA affects PCL/HAp, which behave as a single, integrated system.
However, the asynchronous maps (Figure 8b) exhibit slight differences depending on the molar mass of the polymer. The most visible difference concerns the cross-peaks between the OA bands and the PCL or HAp bands. These cross-peaks are more pronounced in the case of the PCL45k + OA + 15%HAp composite series than in the case of PCL80k + OA + 15%HAp. This observation indicates a well-defined sequence of spectral changes in the case of PCL45k + OA + 15%HAp membranes, initially in the OA band, followed by changes in the PCL or HAp band (Figure S8, Supplemental Materials). In contrast, for PCL80k + OA + 15%HAp series, the corresponding spectral changes seem to occur in a simultaneous rather than sequential manner. This conclusion is also consistent with the synchronous maps analysis, which showed more pronounced cross-peaks for PCL80k + OA + 15%HAp compared to PCL45k + OA + 15%HAp.
Conclusion
The influence of oleic acid on hydroxyapatite-modified poly(ɛ-caprolactone) with composites was tested with respect to the molar mass of PCL. The addition of OA significantly improved the dispersion of HAp in the polymer matrix, as evidenced by visual evaluation, and PLM, SEM, and EDX mapping analysis. When comparing both series of polymer membranes, it was observed that membranes made of PCL45k and OA were very susceptible to cracking, which indicates the destructive effect of OA on the structure of polymeric matrix. The addition of HAp showed stabilization effect through binding with OA. However, such an effect was observed up to a limiting ratio of 3:1 (HAp:OA by weight), above which the membrane was destroyed again. In contrast, the PCL80k series of membranes were more flexible and did not crack. These series were subjected to mechanical tests, which revealed that the addition of HAp improves the mechanical strength of the composites (increase in the Young’s modulus values). However, the increasing concentration of OA ultimately led to gradual worsening of mechanical properties, which turned out to be connected with a decreasing level of polymer crystallinity as revealed by Raman spectroscopic parameters. 2D-COS analysis shows that the effect of OA is different depending on the molar mass of PCL. The response of the composite components to the increasing OA concentration in the case of PCL45k is sequential, initially occurring in the OA band, followed by changes in PCL along with HAp. Whereas, in the case of the PCL80k series, changes occur more simultaneously for all system components. Taken together, the results indicate that the optimal OA content in the PCL/HAp composites, ensuring homogeneous HAp distribution and the highest mechanical performance, corresponds to an OA to HAp weight ratio of 1:6.
Supplemental Material
sj-docx-1-asp-10.1177_00037028261459127 - Supplemental material for Evaluation of Oleic Acid-Modified Hydroxyapatite Polymer Nanocomposites for Regenerative Medicine Applications
Supplemental material, sj-docx-1-asp-10.1177_00037028261459127 for Evaluation of Oleic Acid-Modified Hydroxyapatite Polymer Nanocomposites for Regenerative Medicine Applications by Anna Kołodziej, Anna Magiera1, Małgorzata Świętek, Miroslav Slouf, Jiří Hodan, Daniel Horák, and Aleksandra Wesełucha-Birczyńska in Applied Spectroscopy
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The study was supported by the research part of the subsidy of the Faculty of Chemistry Jagiellonian University in Krakow, Poland. The publication has been supported by a grant from the Faculty of Chemistry under the Strategic Programme Excellence Initiative (Young Laboratories Program, Editions 4 and 5) at Jagiellonian University. This work was partially funded by the National Science Centre, Poland (Project 2024/08/X/ST4/01426 to A.K.). The study was carried out using research infrastructure funded by the European Union in the framework of the Smart Growth Operational Programme, Measure 4.2; Grant no. POIR.04.02.00-00-D001/20, “ATOMIN 2.0–Centre for Materials Research on ATOMic Scale for the INnovative Economy”.
ORCID iDs
Supplemental Material
All supplemental material mentioned in the text accompanies this paper online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.