
Abstract
Familial hypercholesterolaemia, one of the most common inherited diseases in the general population, is associated with mutations in at least three different genes including the low density lipoprotein receptor (LDLR), apolipoprotein B (APOB) and protein convertase subtilisin/kexin type 9 (PCSK9) genes. In this report, we describe an unclassified DNA variant (c.1813C>T; p.Leu605Leu) within exon 12 of the LDLR gene in a kindred in which familial hypercholesterolaemia is associated with c.1813C>T heterozygosity. In silico analysis suggested that c.1813C>T might affect splicing of the LDLR gene by creating a cryptic donor splice site, which was confirmed by RT-PCR coupled with cDNA sequencing, to result in the loss of 34 base pairs in the coding sequence. The latter truncated mRNA is predicted to generate a frameshift leading to a premature stop at codon 652 and early termination of the low density lipoprotein receptor polypeptide chain, and thus provides a molecular basis for the hypercholesterolaemic phenotype. This case report highlights the emerging utility of RNA studies for the molecular diagnosis of familial hypercholesterolaemia in patients with potential mRNA splicing variants.
Background
Hypercholesterolaemia is a major risk factor for atherosclerosis and its cardiovascular complications. Familial hypercholesterolaemia (FH; OMIM#143890), usually an autosomal dominant condition, is characterized by very high plasma concentrations of cholesterol and LDL cholesterol, tendon xanthomas and increased risk of premature coronary heart disease. Mutations in at least three genes have been identified to be associated with FH, including the low density lipoprotein receptor (LDLR), apolipoprotein B (APOB) and protein convertase subtilisin/kexin type 9 (PCSK9) genes. 1 Recently, mutations in the signal transducing adaptor family member 1 (STAP1) gene were also found in patients with FH. 2
The majority of FH cases are associated with mutations in the LDLR gene, which is located in the short arm of chromosome 19 and consists of 18 exons. The LDL receptor contains 839 amino acids in its mature form and mediates the specific uptake of plasma LDL particles into the liver and other tissues of the body. 3 Heterozygotes with one defective LDLR gene typically have markedly raised plasma LDL cholesterol concentrations associated with the occurrence of tendon xanthomas, accelerated atherosclerosis and premature coronary artery disease. Homozygous individuals are more severely affected and may die from coronary artery disease before reaching the age of maturity. The prevalence of heterozygous FH is estimated to be between 0.2 and 0.5% in European and North American populations.4,5 To date, over 1200 variants of the LDLR gene have been identified in patients with FH, including single nucleotide mutations in exons, introns and the promoter region, as well as other DNA rearrangements. 6 In this report, an unclassified variant (a synonymous single nucleotide substitution) of the LDLR gene in an extended family with hypercholesterolaemia is described.
Case report and family history
The proband (II.3; Figure 1), a Caucasian male, was initially referred to the lipid clinic at the age of 38 years after his brother’s fatal heart attack in his early 40s. At the time of presentation, the proband was asymptomatic with an unremarkable past medical history. On examination, he was found to have bilateral corneal arcus and thickening was detectable in his Achilles tendons. Blood pressure was 118/76 mmHg; 12-lead electrocardiography was unremarkable before and after a vigorous exercise tolerance test. His initial fasting serum total cholesterol concentration was 12.8 mmol/L with HDL-C, triglycerides (TG) and LDL-C concentrations of 1.1, 1.2 and 11.2 mmol/L, respectively. A clinical diagnosis of FH was made on the basis of the Simon Broome criteria, which are commonly used in the UK for the management of patients suspected to have FH.
7
His best lipid profile results to date (total cholesterol 5.3, HDL-C 1.1, TG 1.3 and LDL-C 3.6 mmol/L) were achieved using a combination of rosuvastatin 40 mg, ezetimibe 10 mg and colestyramine 12 g daily. Very little is known about the proband’s parents except that his mother (I.2) died of myocardial infarction at the age of 75 years, whereas the age and cause of his father’s death are unknown. The proband’s only sibling (II.1; Figure 1) had his first ischemic heart attack at the age of 43 years and died approximately a year later as a result of a second episode of myocardial infarction. Mutation analysis was not carried out for the proband’s brother. Unfortunately, the latter’s medical records are not accessible; his lipid profiles and treatment are also unknown to the existing family members. He was survived by two daughters (Figure 1). The proband’s elder niece (III.2) was found to have a serum total cholesterol concentration of 7.9 mmol/L at the age of 26 years but no detailed lipid profile was performed before commencement of her lipid-lowering treatment. At the time of writing, she was 43 years old and well. The proband’s grandniece (IV.1) was also found to have relatively high fasting cholesterol concentrations at the age of 12 years (total cholesterol 7.3 mmol/L, HDL-C 1.5 mmol/L, TG 1.3 mmol/L and LDL-C 5.2 mmol/L). The proband’s younger niece (III.3) has declined testing of her cholesterol concentrations and genetic mutation. Lipid profiles of other family members are not available.
Kindred with c.1813C>T variant in the low density lipoprotein receptor (LDLR) gene. The heterozygous variant was detected in three family members (II.3, III.2 and IV.1) who had marked hypercholesterolaemia before lipid-lowering treatment. HDL-C: serum high density lipoprotein cholesterol concentration; LDL-C: serum low density lipoprotein cholesterol concentration; MI: myocardial infarction; TChol: serum total cholesterol concentration; TG: serum triglycerides concentration.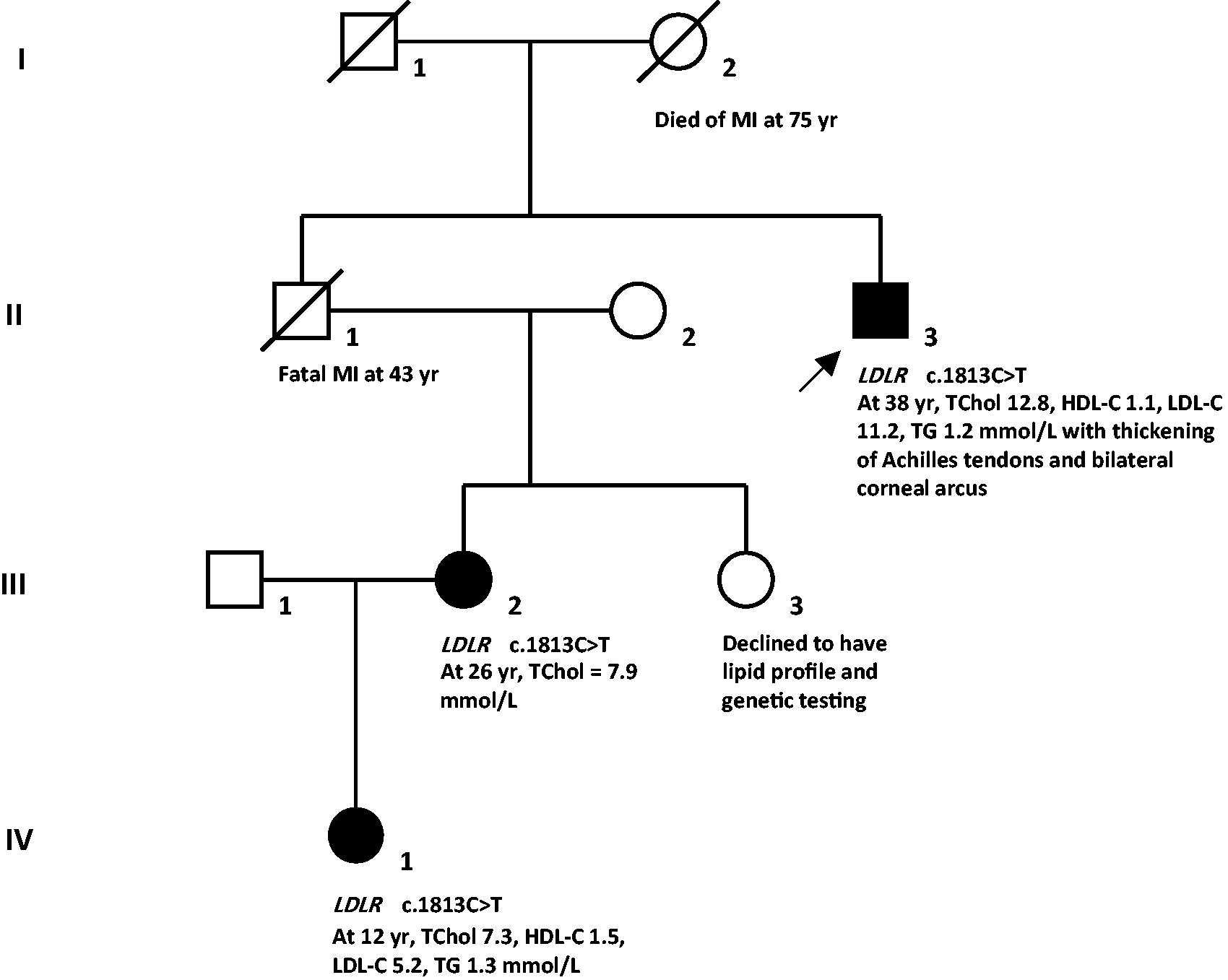
Peripheral blood samples were collected from patients with informed consent. Materials and methods are available as supplementary information on the journal’s website. Sequence analysis of all exons of the LDLR gene using genomic DNA extracted from theproband (II.3) identified a heterozygous single nucleotide change at position 1813 within exon 12(c.1813C>T; p.Leu605Leu). This heterozygous nucleotide change was also detected in genomic DNA from the elder niece (III.2) and grandniece (IV.1) (Figure 1). Although the c.1813C>T substitution is not predicted to alter the amino acid (leucine) at codon 605, in silico analysis suggested that the substitution might affect splicing of the LDLR gene by creating a cryptic donor splice site. Multiplex ligation-dependent probe amplification analysis found no evidence of any large duplication or deletion in the LDLR gene. Direct sequence analysis of genomic DNA from the three patients (proband, III.2 and IV.1) did not detect any mutation commonly found in codon 3527 of the APOB gene or codon 374 of the PCSK9 gene.
Given the above synonymous mutation and potential cryptic donor splice site, total RNA was extracted from the proband’s peripheral blood and reverse transcribed. Subsequent analysis of cDNA confirmed that the sequence variant c.1813C>T resulted in the creation of a donor splice site, leading to the loss of 34 base pairs from exon 12 in the coding sequence (Figure 2). The latter truncated mRNA is predicted to cause a frameshift leading to a premature stop at codon 652 and thus early termination of LDLR translation.
Sanger sequence analysis of cDNA from control (upper panel) and proband (lower panel) showing the heterozygous cDNA variant as a result of the loss of 34 bases from exon 12 in the LDLR gene coding sequence.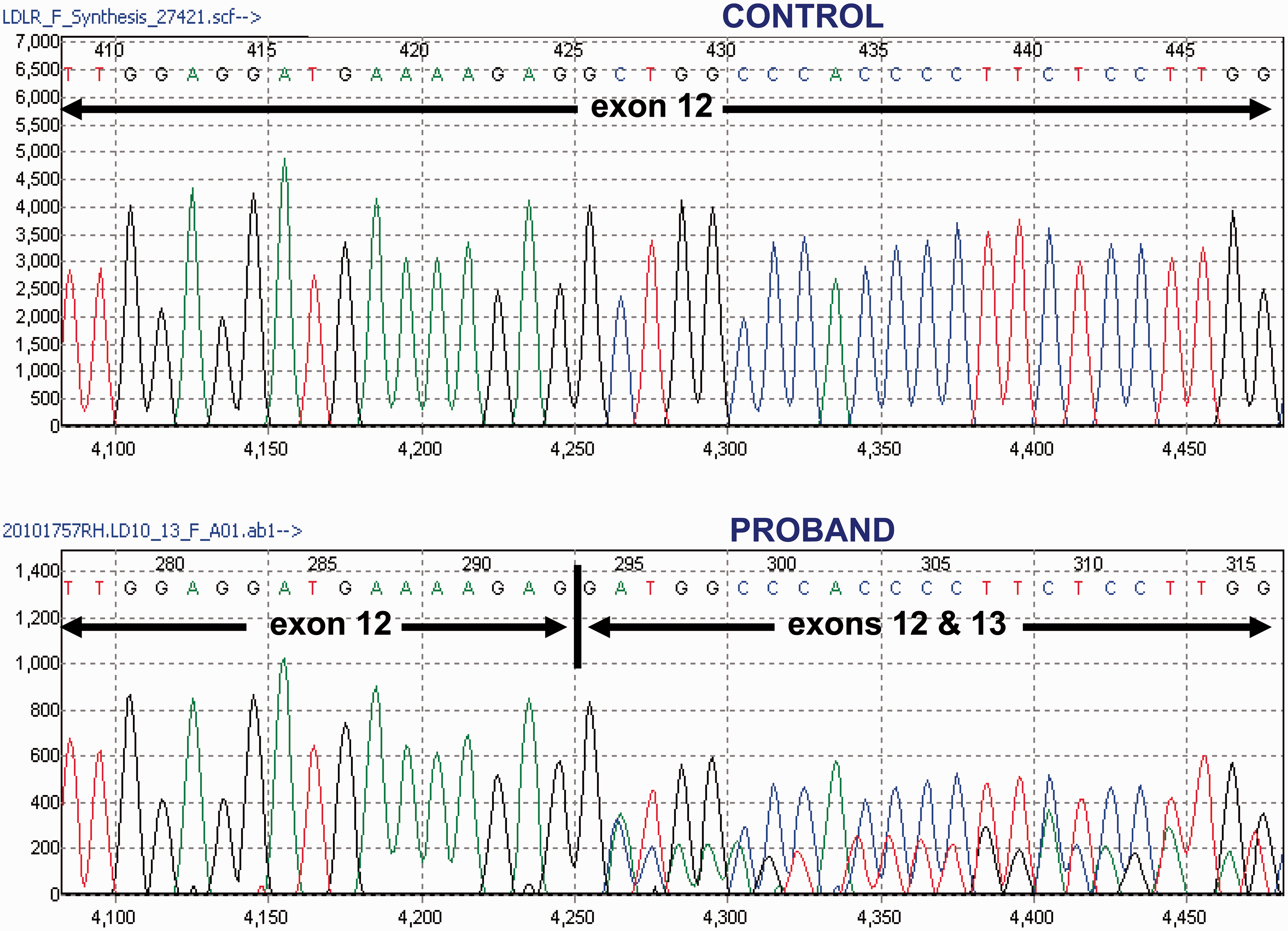
Discussion
To our knowledge, the LDLR gene mutation c.1813C>T has only been briefly reported in one previous study; four members with hypercholesterolaemia belonging to a family in France carried the c.1813C>T mutation, whereas a fifth family member with no hypercholesterolaemia did not. 8 Although RNA analysis was not performed in the French study, genotype–phenotype correlation within that family with FH lends support to a causal relationship between the c.1813C>T variant and hypercholesterolaemia.
In the present study, the proband’s only sibling (II.1) died at 43 years of age due to an ischaemic heart event with an unknown lipid phenotype. Since the latter’s daughter (III.2) and brother (proband; II.3) carry the same c.1813C>T variant and were hypercholesterolaemic before treatment, it is highly probable that he also had the same variant with consequent hypercholesterolaemia, contributing to his premature ischaemic heart disease. However, severe hypercholesterolaemia caused by the heterozygous c.1813C>T mutation might not have been the only factor leading to the premature ischaemic heart disease and death of the proband’s brother. It is possible that, in addition to hypercholesterolaemia, other cardiovascular risk factors such as cigarette smoking, hypertension, diet and obesity might have contributed to the latter’s premature ischaemic heart disease. He might also have mutations in other gene(s) that can increase the risk of myocardial infarction. 9
Compared to the other family members who are also known to be LDLR c.1813C>T heterozygotes, the proband appears to have markedly higher pretreatment serum total cholesterol and LDL-C concentrations. Lifestyle factors, e.g. diet and obesity, and other unidentified mutations within the APOB, PCSK9 or the recently reported STAP1 genes may have contributed to the significantly worse pretreatment lipid profile in the proband.
The present report also highlights the importance of RNA studies for patients who are suspected of carrying genetic mutations that may affect mRNA splicing and thus subsequent protein translation. In recent years, isolation of stable and high-quality RNA from patients’ peripheral blood has become clinically feasible because of the availability of improved technology for blood collection and RNA extraction. Blood collection tubes suitable for RNA transport at room temperature with minimal RNA degradation are now widely available. It is anticipated that RNA studies will become increasingly used in routine clinical lipidology service and clarify the significance of some unclassified variants for the molecular diagnosis ofFH.
In conclusion, in this report we have identified a previously unclassified variant of the LDLR gene associated with hypercholesterolaemia in an extended family. Alternative mRNA splicing was confirmed, leading to premature termination of the polypeptide chain. The importance and increasing availability of RNA studies for the routine molecular diagnosis of FH is also highlighted.
Footnotes
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Informed consent was obtained from the patients. No ethical approval was required.
Guarantor
SWW.
Contributorship
CKMH wrote and edited the manuscript. FRM wrote the first draft of the manuscript. CB performed the molecular assays. SWW conceived the study. All authors reviewed the manuscript and approved the final version.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.