
Abstract

Introduction
Low phospholipid-associated cholelithiasis (LPAC) syndrome is primarily characterized by the development of symptomatic recurrent microlithiasis and cholelithiasis in early adulthood (<40 years of age). This syndrome is rarely diagnosed in children or adolescents. Other manifestations of LPAC include cholecystitis, cholangitis, intrahepatic gallstones, acute recurrent biliary pancreatitis, and recurrence of biliary symptoms after cholecystectomy.1,2 Over time, biliary fibrosis or cirrhosis may develop.3,4
LPAC is associated with gene sequence variants in the adenosine triphosphate-binding cassette, subfamily B, member (ABCB4) gene. Mutations are mostly heterozygous (monoallelic) frame-shift, nonsense or missense; rarely biallelic missense mutations have been reported. 3 In addition to LPAC syndrome, ABCB4 gene variants are involved in several other hepatobiliary disorders, including progressive familial intrahepatic cholestasis (PFIC3), transient neonatal cholestasis, intrahepatic cholestasis of pregnancy (ICP), drug-induced cholestasis, primary sclerosing cholangitis, and primary biliary cirrhosis.3-7 ABCB4 encodes the multidrug resistance protein 3 (MDR3). MDR3 is located in the hepatocyte canalicular membrane and is involved in the transport of phosphatidylcholine (PC) into bile. PC within the bile is essential for appropriate solubilization of cholesterol via micelle and cholesterol-phophatidylcholine vesicle formation. Phosphatidylcholine, also, acts within the biliary tract as a cytoprotectant against the damaging effects of bile salts. Thus, the pathophysiological basis of LPAC syndrome is a decrease in the secretion of PC into the bile. The resultant phospholipid-deficient bile leads to biliary cholesterol supersaturation (ie, increase in the biliary cholesterol-to-phospholipid ratio) with subsequent increased bile lithogenicity and formation of cholesterol-rich stones.2-4,6,7 A summary of the main features of LPAC syndrome is presented in Table 1.
Features of LPAC Syndrome
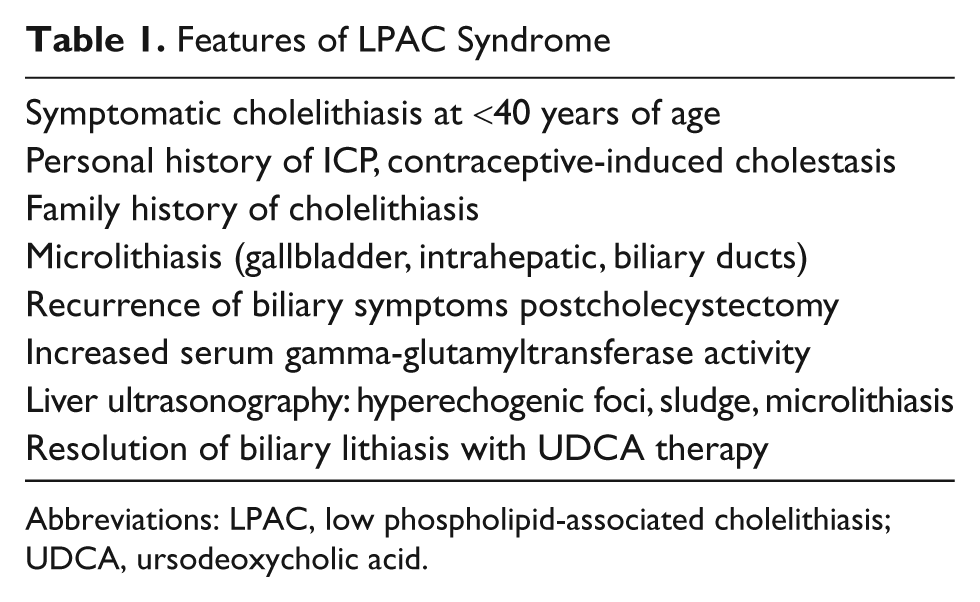
Abbreviations: LPAC, low phospholipid-associated cholelithiasis; UDCA, ursodeoxycholic acid.
Case Report
A 15-year-old male presented with a 24-hour history of epigastric pain, nausea, and vomiting. This was the first reported episode of significant abdominal pain. The patient’s body mass index was 21.2 at the time of presentation. His past medical history was negative for previous hospital admissions, anemia, jaundice, and prolonged illnesses. The patient was adopted during early childhood. His family medical history is limited to knowing his mother had liver disease of unknown etiology and a second maternal cousin developed cholelithiasis at 26 years of age. The patient was diagnosed with cholecystitis, cholelithiasis, choledocholithiasis, and pancreatitis based on his clinical presentation, laboratory (Table 2), abdominal ultrasound, and abdominal computed tomography scan findings. Electrolytes, hemoglobin, hematocrit, platelets count, and serum triglyceride level were all normal. He had a mild coagulopathy. Two days after admission, he underwent endoscopic retrograde cholangiopancreatography (ERCP). Numerous small cholesterol stones were found in the common bile duct (CBD) and removed (Figure 1). A sphincterotomy was performed and a stent placed. His amylase and lipase improved after the ERCP. His prothrombin time and international normalized ratio abnormalities corrected with oral vitamin K. One week after presentation, he underwent laparoscopic cholecystectomy. The gallbladder wall was thickened (up to 0.5 cm), and the mucosal surface was diffusely eroded and hemorrhagic. The lumen contained multiple yellow (cholesterol-like) multifaceted calculi ranging from 0.1 to 0.5 cm in diameter. Three weeks after the cholecystectomy, a magnetic resonance cholangiopancreatography (MRCP) revealed CBD dilatation (12 mm) with numerous intraductal opacities measuring less than 1 cm in diameter within the CBD (Figure 1). Also, 3 regions within the pancreas had magnetic resonance characteristics of pseudocysts. A second ERCP confirmed the presence of CBD dilation and multiple intraductal stones (Figure 1). The previously placed stent and all stones were removed with dilatation of the previous sphincterotomy. Mutational analysis of the ABCB4 gene performed by a contract laboratory revealed a heterozygous novel gene variant, c.1685C >T (p.A562V). The patient was placed on ursodeoxycholic acid (UDCA; 10 mg/kg/d). Two months after initiating this therapy, he redeveloped persistent epigastric abdominal pain. The pain resolved after an increase in the UDCA to 15 mg/kg/d. Four months after presentation, a follow-up MRCP revealed improvement in the CBD dilatation (9 mm), no evidence of cholelithiasis, and resolution of 2 of the pancreatic pseudocysts.
Laboratory Values
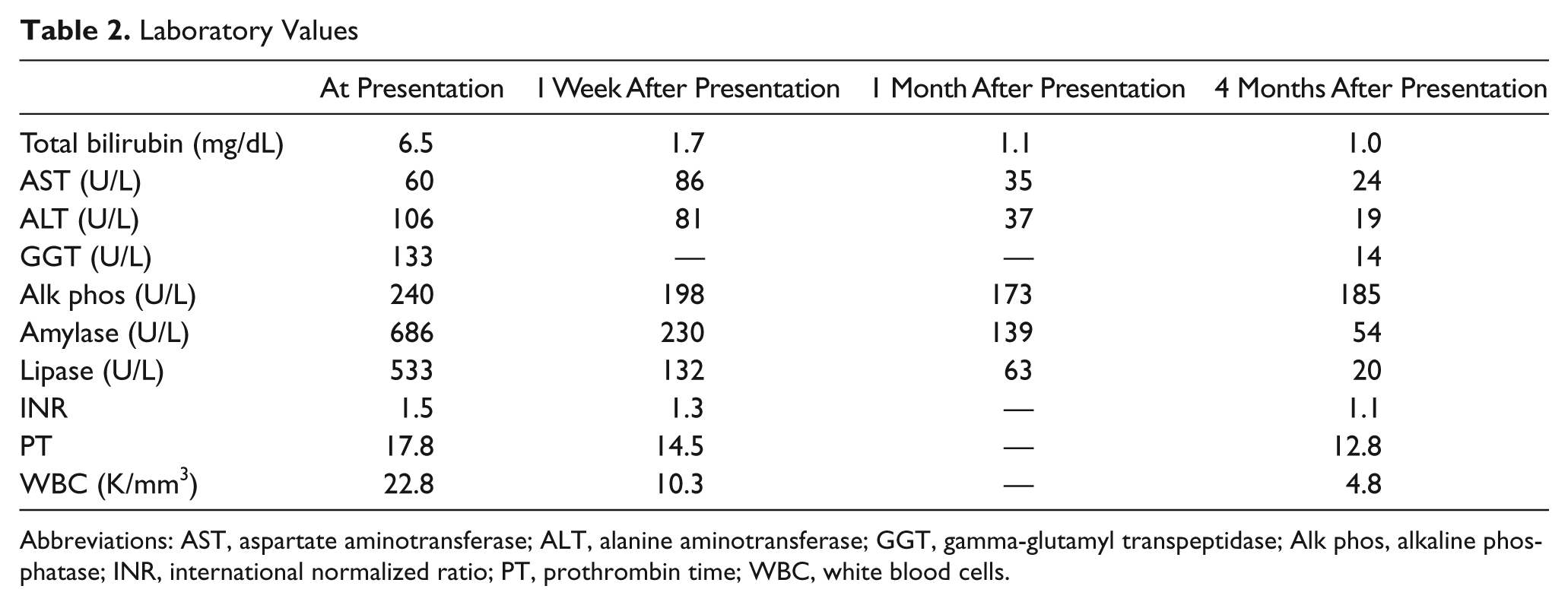
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transpeptidase; Alk phos, alkaline phosphatase; INR, international normalized ratio; PT, prothrombin time; WBC, white blood cells.
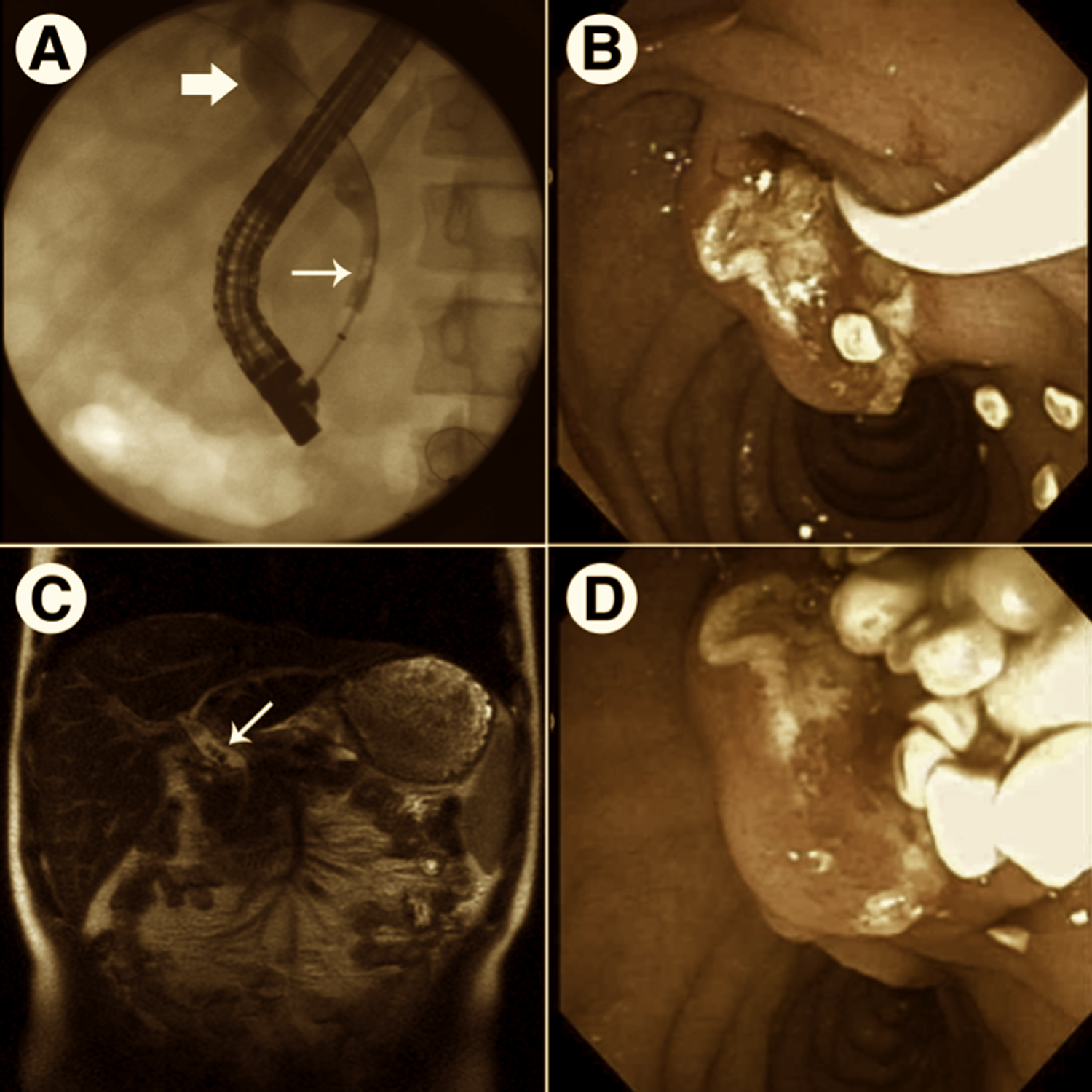
Radiographic and endoscopic images of biliary abnormalities observed in the patient with LPAC syndrome (A and B) Obtained at diagnosis. (C and D) Obtained 4 weeks after initial ERCP, postcholecystectomy. (A) Fluoroscopic image obtained during ERCP demonstrating numerous filling defects (small arrow) within and proximal dilation (large arrow) of the common bile duct. (B and D) Endoscopic images showing multiple pale cholesterol-like stones being removed through the ampulla. (C) MRCP image demonstrating filling defects (small arrow) within the common bile duct.
Discussion
Presentation during early adulthood is one of the characteristic features of LPAC syndrome; however, diagnosis during childhood and adolescence is quite uncommon. In fact, our patient is one of the youngest patients diagnosed with this syndrome to be reported. His diagnosis was based on clinical presentation (microlithiasis, elevated gamma-glutamyltransferase [GGT], and a possible family history of early onset cholelithiasis) and the finding of a missense genetic variant in the ABCB4 gene. Though this variant has not been identified previously in patients with LPAC, the resulting amino acid change is predicted to be deleterious to the function of the protein by SIFT and PolyPhen algorithms.
Previous case studies have documented that LPAC can present during adolescence. For example, our search of the literature identified 2 cases describing the presentation of symptoms associated with LPAC syndrome in adolescent patients in which the diagnosis was made years after this early initial presentation. Both patients were females with a history of early onset of biliary symptoms.4,8 In one case, the patient was diagnosed with LPAC syndrome at 47 years of age after presenting with chronic cholestasis and recurrent ascites secondary to biliary cirrhosis. 4 Her past medical history was significant for having undergone a cholecystectomy for symptomtic cholelithiasis at 19 years of age. In the second case, the patient was diagnosed with LPAC syndrome after her 38-year-old father was diagnosed with the syndrome. 8 She had undergone a cholecystectomy for acute cholecystitis and cholelithiasis at 13 years of age.
In all cases where genotyping confirms the diagnosis or the clinical presentation is highly suggestive of LPAC syndrome, long-term therapy with ursodeoxycholic acid (UDCA) should be initiated. UDCA enhances bile flow, inhibits the toxicity of endogenous hydrophobic bile acids, and increases the pool of protective hydrophilic bile acids. Empiric evidence suggests that UDCA prevents the recurrence of cholelithiasis and leads to an improvement in the signs and symptoms of this syndrome. Denk et al reported that relatively high doses of UDCA (20 mg/kg/d) resulted in the improvement of liver tests (aspartate aminotransferase, alanine aminotransferase, and GGT). 8 We observed an improvement in abdominal pain in our patient after an increase in the UDCA dose to 15 mg/kg/d. This improvement might reflect the fact that biliary pain in LPAC can be due to cholesterol crystal deposits and biliary inflammation rather than to overt cholelithiasis. Cholecystectomy is indicated for symptomatic gallstones, but not microlithiasis or sludge within the gallbladder as this might respond to UDCA. Biliary drainage or partial hepatectomy may be indicated for symptomatic intrahepatic bile duct dilation with gallstones.2-6
Although LPAC syndrome is an infrequent cause of cholelithiasis in all age groups, it should be suspected in adolescent patients with symptomatic cholelithiasis, intermittent cholestatic attacks, sludge in the intrahepatic bile ducts, or recurrence of symptoms after cholecystectomy. Molecular diagnosis can be made by analyses of ABCB4 gene. Early diagnosis of LPAC syndrome is important since the clinical course is progressive, with multiple recurrences of cholelithiasis, bile duct dilatations, biliary fibrosis, or cirrhosis despite operative intervention(s) until UDCA treatment is started. UDCA appears to prevent or improve the symptoms and complications of this syndrome. From limited data, patients with LPAC syndrome might benefit from higher than the usual recommended doses of UDCA (15-20 mg/kg/d).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.