
Abstract
Objective. Asymmetric crying facies (ACF) is congenital hypoplasia of the depressor anguli oris muscle characterized by asymmetry of lower lip depression during crying. This has an overall incidence of 0.6%. This study determines the incidence of ACF in a large population of patients with 22q11.2 deletion. Patients and Methods. A retrospective review of medical records on patients with a confirmed 22q11.2 deletion was undertaken. Results. A total of 836 records were reviewed. Of these, 117 (14%) were noted to have ACF on physical examination. Within this latter group, palatal anomalies were common (77%), as was congenital heart disease (78%); however, these numbers did not differ significantly from their known prevalence in the 22q11.2 population. Conclusions. We report a 14% incidence of ACF in patients with a 22q11.2 deletion, significantly higher than in the general population. We suggest, therefore, that newborns with ACF be referred for further screening for the 22q11.2 deletion syndrome.
Introduction
Asymmetric crying facies (ACF) is recognized in neonates as lower lip asymmetry present only with crying. The typical clinical picture is significant unilateral depression of the lower lip during crying noted immediately postpartum, while forehead wrinkling, nasolabial fold depth, and eyelid closure remain bilaterally intact (Figure 1). 1 Later on in infancy, mild asymmetry of the lower lip is often noted with a full open mouth smile. The incidence of ACF in the general population is about 0.6%.1-6
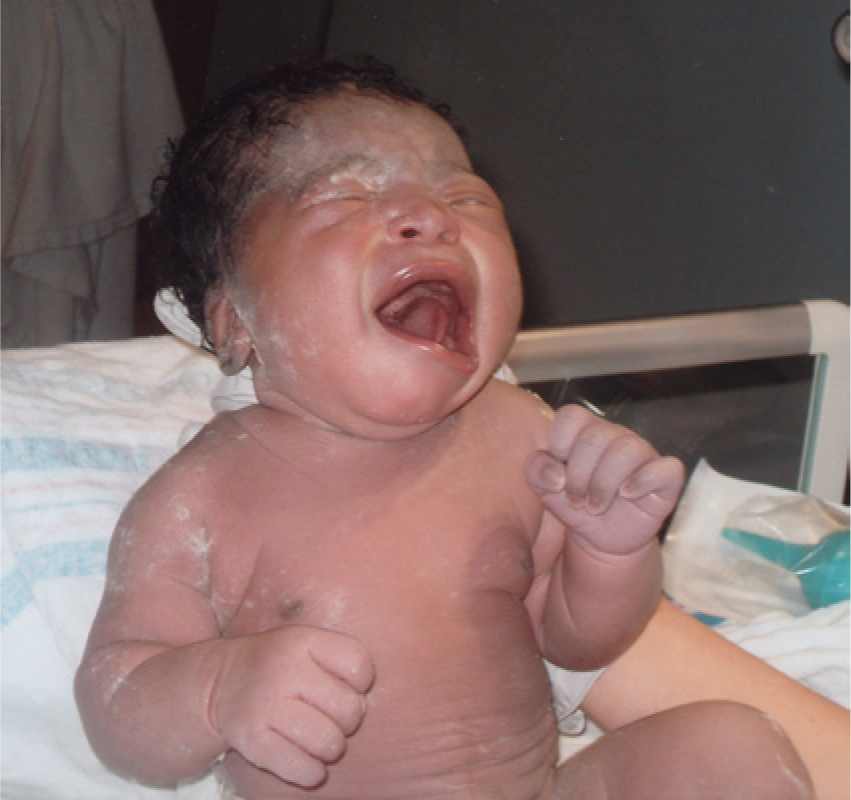
Asymmetric crying facies apparent at the time of birth.
Asymmetric crying facies occurs as a result of unilateral insufficient downward motion of the lip. This unilateral weakness leads to exaggerated lower lip depression on the normal side, exacerbating the asymmetry. Downward movement of the lower lip is produced by the depressor anguli oris and depressor labii inferioris muscles, which are both innervated by marginal mandibular branches of the facial nerve (Figures 2 and 3). 2
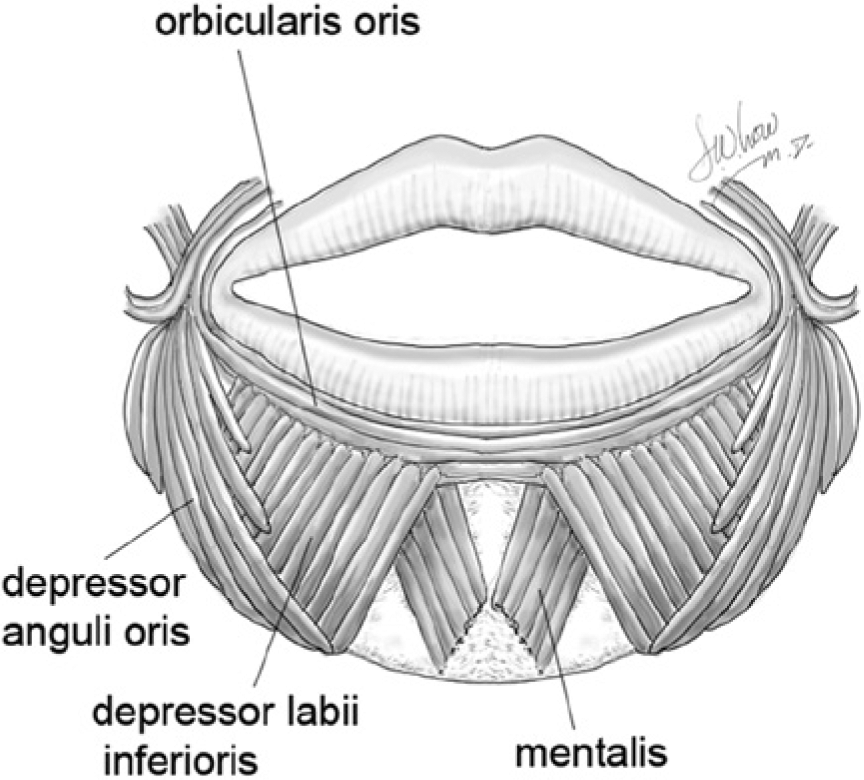
The anatomy of the lower lip depressor muscles.
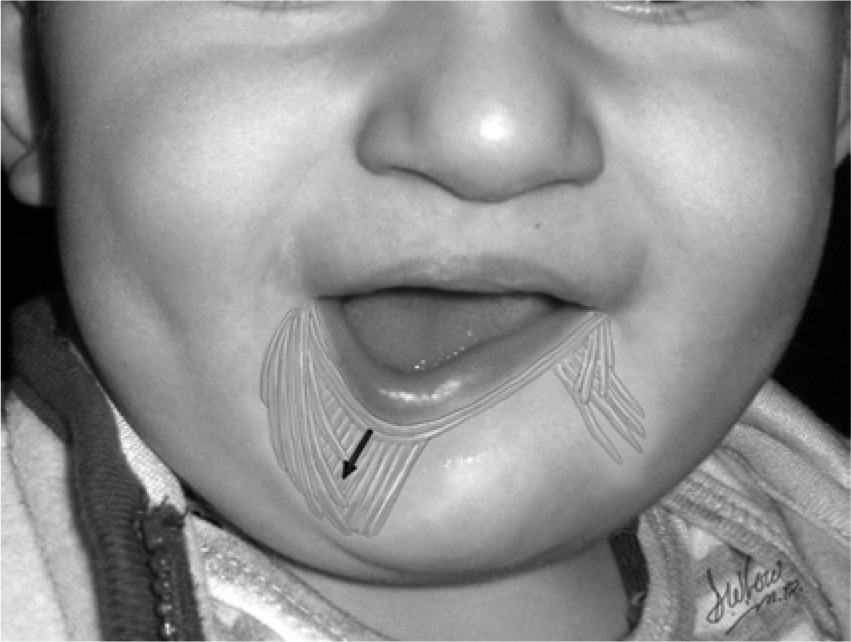
A depiction of the unilateral deficiency of depressor labii inferioris, resulting in unopposed lower lip depression on the intact side and an asymmetric smile.
The etiology of ACF can be differentiated as secondary to either facial nerve compression or faulty facial muscle/nerve development. 7 While facial nerve compression often spontaneously resolves with no residual deficit, the latter developmental defects generally do not resolve over time. Electrodiagnostic testing can serve to differentiate the 2 etiologies in infants with ACF, revealing that approximately 20% of infants with ACF have a nerve compression etiology and 80% of patients have developmental abnormalities resulting in muscle hypoplasia. 8
Nerve compression as an etiology for ACF in infancy is common as the facial nerve is easily prone to trauma. The marginal mandibular branch is not protected under the mandible in infants as it is in the adult population, but instead it lies on top of the mandible where it is more exposed to compressive injury in both the antepartum and intrapartum period.2,7 Faulty facial muscle/nerve development, however, is responsible for the vast majority of unresolving ACF. Hypoplasia of the depressor anguli oris or depressor labii inferioris muscles may be primary or may be secondary to insufficient innervation of muscle fibers. Intrauterine molding and viral infection during pregnancy have been implicated as potentially etiologic4,9-11; however, the search for a genetic etiology began when more than half of ACF patients examined were noted to have one or more relatives with the condition. 4 Vertical transmission was proposed and supported.12-14
In patients with ACF secondary to a developmental error in muscle or nerve development, a number of other congenital anomalies may be present. Malformations of all body parts, especially the heart, have been observed, as has neuroblastoma and mediastinal teratoma. Trisomy 18 and neurofibromatosis type I have also been observed as an etiology for ACF. 2 These major malformations occur within the ACF population at a frequency of 5% to 77%. 2 In 5 prospective studies, the range of associated congenital anomalies in the ACF population was 5% to 20%, for a total combined incidence of 9.4% (14/149 newborns).1-6 A known genetic association with ACF is Cayler cardiofacial syndrome, which has been found to be associated with the 22q11.2 deletion in a subset of patients.10,15 The chromosome 22q11.2 deletion is additionally the underlying genetic cause of DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly face syndrome and a subset of patients with Opitz G/BBB syndrome. 1
While 22q11.2 deletion syndrome (22q11DS) is the most common microdeletion syndrome, the presence of ACF in this population has not yet been extensively studied. The incidence of ACF in cases of chromosome 22q11DS has previously been reported from 2% to 33%, although most series are small. 16 The purpose of this study was to determine the prevalence of ACF in a large cohort of patients with a 22q11DS and to ultimately assess whether the diagnosis of ACF in a newborn should prompt screening for the 22q11.2 deletion and other associated features.
Patients and Methods
A retrospective medical record review was undertaken of all patients with a confirmed 22q11.2 deletion followed at the “22q and You” Center at the Children’s Hospital of Philadelphia from 1996 to 2009. The diagnosis was confirmed by fluorescence in situ hybridization or array-based studies. The presence or absence of ACF was determined based on physical examination records. In the patients with documented ACF, demographic data including sex and race were recorded. To help identify perinatal trauma as a potential cause of ACF, mode of delivery and perinatal complications were documented. To identify other congenital anomalies present in the 22q11.2 deletion population with ACF, palatal anomalies, velopharyngeal insufficiency, and congenital heart disease were reviewed from medical chart records.
Results
Records on 836 patients with a documented 22q11.2 deletion were reviewed. Of these, 117 (14%) were noted to have ACF on physical examination. Within this latter group (n = 117), 67 were female (57%) and 50 male (42%). Ethnicity was noted on 116/117 patients including Caucasian (n = 100, 85%), African American (n = 6, 5%), Asian (n = 6, 5%), and Hispanic (n = 4, 5%), consistent with our overall clinic distribution. Mode of delivery was documented on 101 patients and included spontaneous vaginal delivery (n = 64, 63%) and caesarian section (n = 37, 37%). Three patients delivered vaginally had forceps applied (3% of total).
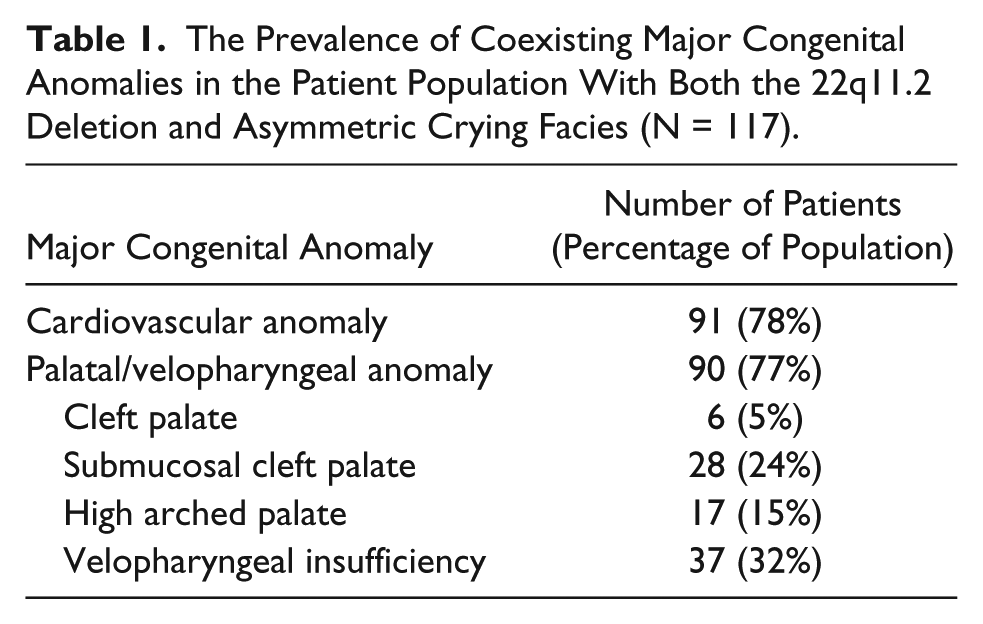
The presence of coexisting congenital anomalies was common and the results are recorded in Table 1. Seventy-eight percent of patients with both the 22q11 deletion and ACF had concurrent cardiovascular anomalies, such as atrial septal defect, ventricular septal defect, and vascular rings. This number did not differ significantly from the population of all patients with 22q11.2 deletion (75%).17,18 Overt cleft palate was present in 6 patients (5%), while a submucosal cleft palate was detected in 28 (24%). Seventeen patients (15%) had a high arched palate, and 37 patients (32%) had documented velopharyngeal insufficiency. While these palatal anomalies and velopharyngeal abnormalities were common (77% of patients), the incidence did not differ significantly from the population of all patients with the 22q11.2 deletion (76%).17,18
The Prevalence of Coexisting Major Congenital Anomalies in the Patient Population With Both the 22q11.2 Deletion and Asymmetric Crying Facies (N = 117).
Discussion
Asymmetric crying facies is most commonly caused by congenital anomalies: either improper marginal mandibular branch development or hypoplasia of the depressor anguli oris or depressor labii inferioris muscles. While these congenital anomalies have previously been thought to be secondary to viral infection during pregnancy or abnormal fetal posturing, there have been genetic implications.2,4,12,13 The 22q11.2 deletion syndrome has previously been implicated in the etiology of congenital ACF.1,16
The 22q11.2 deletion syndrome is the most common microdeletion syndrome with both palatal and cardiovascular involvement. Although suspected to be underrecognized due to its clinical variability, 22q11.2 deletion syndrome has an estimated prevalence of 1:2000 to 4000 live births in the United States.19-21 The classic clinical findings include immunodeficiency (77% of patients with 22q11.2 deletion syndrome), palatal abnormalities (76%), and congenital heart disease (75%). Commonly, endocrine dysfunction (50%), gastrointestinal disorders (35%), renal disease (35%), and developmental delay are also present with 25% developing schizophrenia in adulthood.17,18 The variability in clinical presentation of the 22q11.2 deletion syndrome has resulted historically in the description of different syndromes sharing the same genetic etiology including DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly face syndrome.22 -30 This enormous phenotypic heterogeneity, however, may result in a delayed diagnosis of several years, especially when a congenital heart defect is absent. 22 Atypical or minimal phenotypes may be missed completely. Even associated facial features are variable and may be missed especially in non-Caucasian individuals.19,22 Neonatal screening for the 22q11.2 deletion is also currently under consideration but it will likely take quite some time for implementation in all states within the United States and may not occur at all outside of the United States.
Asymmetric crying facies remains a lesser-known feature of the 22q11.2 deletion syndrome. The incidence of ACF in patients with a 22q11.2 deletion has previously been reported at 2% to 33% in smaller sample sizes.1,16 In an attempt to identify previously undiagnosed cases of 22q11.2 deletion, deletion studies have been suggested when ACF is present. 2 In addition to a work-up for congenital cardiac anomalies, audiometry, spine and chest radiographs, and a renal ultrasound have been suggested for all patients with ACF.2,31
In this large retrospective review, we report a 14% incidence of ACF in patients with a 22q11DS, significantly higher than the general population incidence of 0.6%. 2 The presence of concurrent cardiovascular anomalies and palatal/velopharyngeal abnormalities among patients with both the 22q11.2 deletion and ACF did not differ significantly from that in the population of all patients with 22q11.2 deletion. 19 We suggest, therefore, that all newborns noted to have this subtle facial sign, especially in the presence of congenital heart disease and/or palatal abnormalities (clefting, velopharyngeal insufficiency, and significant feeding difficulty), be referred for further screening for the 22q11.2 deletion syndrome. Likewise, we reinforce the previous recommendations to evaluate for structural anomalies such as congenital heart disease in patients with ACF.
Ultimately, the 22q11.2 deletion as an etiology for ACF implies permanent loss of lip depressor motion. Treatment options for ACF to help improve facial symmetry exist and include botulinim A toxin injections into the contralateral depressor anguli oris muscle for temporary paralysis and reduction of asymmetry.32,33 Permanent strategies include selective myectomy of the contralateral depressor anguli oris muscle and selective neurectomy of the contralateral marginal mandibular branch of the facial nerve. 34 The goal of these treatments is to reduce the cosmetic and functional deficit that results from congenital ACF and improve quality of life, whether or not the patient has a coexisting 22q11.2 deletion syndrome. This is important information for patients and families as many have expressed an erroneous belief that no treatment strategies exist and therefore ACF is a lifelong permanent disfigurement.
The main limitations of this study relate to its retrospective nature. This trial depended solely on physical examination records; thus, some patients with ACF may have potentially been missed from this study. Although information regarding mode of delivery was collected to assess for possible nerve compression etiology, further details about the perinatal period were not available.
Conclusions
In this extensive review, we report a 14% incidence of ACF in patients with a 22q11.2 deletion, significantly higher than in the general population. We suggest, therefore, that all newborns with ACF, with or without cardiovascular or palatal abnormalities, be referred for further screening for the 22q11.2 deletion syndrome. Likewise, we recommend evaluation for such structural anomalies in patients with ACF.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.