
Abstract

Introduction
Monocarboxylate transporter 8 (MCT8) deficiency is an X chromosome–linked devastating neurodevelopmental disorder, resulting from a functional deficiency in cell membrane protein specific for iodothyronine transport, including the active thyroid hormone triiodothyronine (T3) and encoded by the SLC16A2 gene. The prevalence of MCT8 deficiency is more common than previously thought and is likely in the same order of magnitude as creatine transporter deficiency (SLC6A8) or Rett syndrome (MECP2). 1 MCT8 is expressed in endothelial cells of blood vessels in the brain and is responsible for the transfer of T3 across the blood brain barrier (S. Refetoff, unpublished data). MCT8 deficiency is associated with generalized signs of delayed brain maturation, hypomyelination, altered neuronal differentiation, and deficient synaptogenesis. 2 The disorder results in thyroid hormone deficiency in the brain (with neurologic and psychomotor signs and symptoms of severe cognitive impairment, lack of speech, severe hypotonia, and dystonia) and a hypermetabolic state in the rest of the body due to the high serum levels of T3, accessible to peripheral tissues through alternative transporters (with poor weight gain and loss of muscle mass). Hypo- or delayed myelination or other signs of delayed brain maturation seen on magnetic resonance imaging is not specific for MCT8 deficiency but results from comprehensive thyroid testing are specific for MCT8 deficiency. We report a family with multiple affected males diagnosed with cerebral palsy, with extensive and costly medical workup that most likely would have been avoided with early attention to family history and comprehensive thyroid function testing, including T3 and reverse T3. Feasibility and appropriateness of inclusion of this disorder in newborn screening (NBS) programs is briefly discussed.
Case Report
Two half-brothers (8- and 1-year-old, respectively) sharing the same mother were referred to Genetics for evaluation. The older brother (patient 1, Table 1) diagnosed with cerebral palsy (CP) was wheelchair-bound and never walked, and was gastrostomy-tube dependent since 9 months of age because of poor suck and swallow, and poor weight gain. He exhibited severe cognitive impairment, no speech, truncal hypotonia, diminished muscle mass and generalized muscle weakness, dystonia, dyskinesia, and joint contractures. The younger brother was at the fifth percentile for weight and had myopathic facies, poor head control, truncal hypotonia, dystonia of extremities, and no words. Brain magnetic resonance images at similar ages were essentially identical for both brothers with nonspecific mild diffuse parenchymal volume loss or underdevelopment, delayed myelination, and marked thinning of corpus callosum. Laboratory tests on the older brother had included all normal chromosomal microarray analysis, karyoptype analysis, fragile X testing, Prader-Willi syndrome methylation testing, plasma acylcarnitines, creatine kinase, lactate and pyruvate, urine organic acids, and transferrin for congenital disorders of glycosylation. Family history was suggestive for either an X-linked disorder with another male relative (cousin from a maternal half-aunt through the maternal grandmother) also diagnosed with CP, or a mitochondrial DNA-based disorder. Comprehensive mitochondrial DNA sequencing on the younger brother (patient 2, Table 1) was normal. However, an X-linked intellectual disability (XLID) 87-gene panel testing (Ambry Genetics, Aliso Viejo, CA) on patient 2 identified a novel hemizygous frameshift mutation in the SLC16A2 gene (c.321_322delTG), and thus the diagnosis of MCT8 deficiency (also known as Allan-Herndon-Dudley syndrome). Subsequent testing revealed significantly elevated serum-free and total 3,5,3′-triiodothyronine (T3) and low serum 3,3′,5′-triiodothyronine (reverse T3; rT3) for both brothers, biochemically confirming the diagnosis (Table 1). Serum-free and total thyroxine (T4) were below or at the low level of normal but thyroid-stimulating hormone (TSH) concentrations were within reference ranges (Table 1).
Comprehensive Thyroid Function Testing on the Half-Brothers and Their Mother.
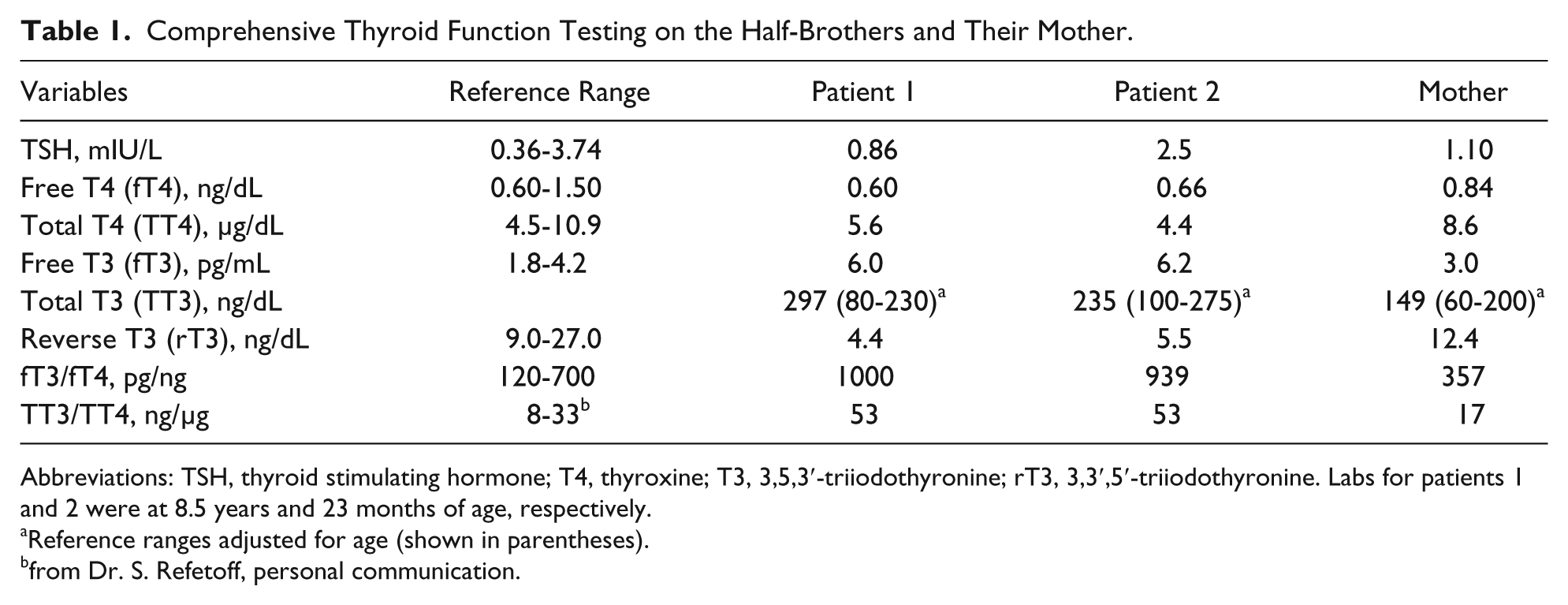
Abbreviations: TSH, thyroid stimulating hormone; T4, thyroxine; T3, 3,5,3′-triiodothyronine; rT3, 3,3′,5′-triiodothyronine. Labs for patients 1 and 2 were at 8.5 years and 23 months of age, respectively.
Reference ranges adjusted for age (shown in parentheses).
from Dr. S. Refetoff, personal communication.
Discussion
Clinical Suspicion and Burden of First Testing
Pediatricians are usually the first to evaluate an infant with hypotonia, head lag, or poor weight gain (the early signs and symptoms of this disorder, but not specific for MCT8 deficiency), and this usually prompts thyroid function testing. However, screening for a thyroid disorder most commonly includes determination of free T4 and TSH concentrations only, 3 and not T3 (free or total) or rT3. Serum T4 concentration in MCT8 deficiency is often reduced, but may be within the low normal range, while serum TSH concentrations are usually normal or slightly elevated. Thus, normal free T4 and TSH concentrations could be falsely reassuring, and a thyroid function abnormality could be dismissed. Consequently, extensive and costly workup for other genetic, genomic or inborn errors of metabolism is most likely pursued, delaying the correct diagnosis, genetic counseling to affected families, and initiation of available therapy for this disorder. Therefore, the clinical suspicion and burden of first testing for this disorder lies with the pediatrician.
Therapies for MCT8 Deficiency
Experience and understanding of the efficacy of therapy and proper management in individuals with MCT8 deficiency remain limited.4,5 Current therapies ameliorate the peripheral (non–central nervous system) tissue thyrotoxicity in this disorder but seem to result in no significant neurologic improvement.
4
Thyroid hormone treatment alone, during childhood has no beneficial effect, and can exacerbate the high serum T3 levels and the resulting peripheral tissue hypermetabolic state.
6
However, combination therapy using high-dose levothyroxine (
Consideration for Inclusion of MCT8 Deficiency in Newborn Screening
In congenital hypothyroidism (CH) thyroid hormone deficiency starts around birth, due to the degree of protection afforded by the maternal thyroid hormone, and so early identification (through NBS) and intervention results in sparing of brain damage. In MCT8 deficiency, however, insult to the brain occurs before the 30th gestational week and thus early identification and possible therapeutic intervention becomes crucial. 2 Based on mice deficient in Mct8, low rT3 is the first abnormality observed after birth and may be the first abnormality noted in humans. 9 Reverse T3 is normally high in infants and in subjects with nonthyroidal illness; thus a low rT3 in any neonate or infant should be concerning and trigger an endocrine and genetic evaluation. Newborn screening programs do not currently test for T3 or rT3 concentrations in dried blood spots (DBSs). The electrospray ionization tandem mass spectrometry (ESI MS/MS) technology, used in most NBS state laboratories for screening certain organic acidemias and fatty acid oxidation disorders, has been used for identification and quantification of mixtures of T3 and rT3 isomers without prior chromatographic separation. 10 Thus, adding T3 and rT3 testing to existing screening by ESI MS/MS may not impose a large added cost or require additional personnel or equipment. Serious discussion about the inclusion of thyroid hormone transporter deficiency screening in NBS (Table 2) is needed.
Inborn Errors of Metabolism, Endocrinopathies, and Some Neuropathies With Neonatal and/or Infantile Hypotonia Not Currently Identified by Newborn Screening. a

Implementation of glycogenosis type II (Pompe disease) testing as well as testing certain lysosomal storage diseases with neonatal/infantile presentation of hypotonia, such as Niemann-Pick disease (A/B), in newborn screening programs are under development.
Conclusion
MCT8 deficiency should be high on the differential diagnosis in the evaluation of a floppy male infant with developmental delay.7,11 Enhanced awareness of the proper selection of thyroid function screening tests, including T3 and rT3, at the earliest signs and symptoms of this disorder, attention to family history (where males are affected most exclusively), and inclusion of this disorder in newborn screening, would lend to early diagnosis, therapeutic intervention, and proper genetic counseling for this devastating disorder.
Author Contributions
JKB wrote the first draft. All authors contributed to the acquisition, analysis and interpretation of data as well as in revising this article critically. JKB, TT, STG, and NEB were involved in the care of the reported subjects.
Footnotes
Acknowledgements
We thank Drs. Samuel Refetoff and Douglas Kerr for discussion and critically reading this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.