
Abstract

Introduction
Seizures are the most common pediatric neurologic disorder. Approximately 30 000 among the 150 000 children, who will sustain a first-time, unprovoked seizure each year, will develop epilepsy. The incidence of epilepsy is highest among children younger than 3 years of age. Approximately 40% of the seizures in this population it is secondary to epileptic encephalopathies. 1
Epileptic encephalopathies (EE) are a heterogeneous group of epileptic disorders that occur early in life. Epileptic Encephalopathies have been classified into various syndromes depending upon the age of onset that include early infantile epileptic encephalopathy (EIEE)/Ohtahara syndrome (OS), Infantile spasm (West syndrome), late infantile epileptic encephalopathy, Dravet syndrome, Early myoclonic encephalopathy, Malignant migrating partial seizures of infancy and Lennox-Gastaut syndrome. It has been postulated that some of these syndromes may represent a spectrum of diseases along a continuum, which may evolve overtime with maturation of the nervous system 2 . To date, 28 types of EIEE have been reported based on the de novo mutations (http://www.omim.org). These syndromes are characterized by intractable seizures resistant to anti-convulsant treatment, persistent severe electroencephalographic abnormalities, and progressive cognitive dysfunction. Epileptic encephalopathies are commonly due to sporadic, de novo dominant mutations in a single autosomal gene. However, autosomal recessive and X-linked recessive forms have also been reported. 2 Advances in the genetic testing methods like whole exome sequencing (WES) and whole genome CNV (copy number variants) sequencing (WGS) has led to identification of newer sporadic de novo autosomal mutations causing epileptic encephalopathies.
Hitherto, sporadic de novo heterogeneous mutations in voltage gated sodium channel (VGSC) [SCN2A] and N – Methyl D – Aspartate (NMDA) receptor [Glutamate receptor ionotropic NMDA2A] (GRIN2A) genes occurring individually have been reported to cause intractable pharmacoresistant seizures2-4. However, these two mutations occurring simultaneously in a phenotypically normal neonate with structurally normal brain presenting with intermittent clusters of intractable seizures associated with ictal vocalizations have not been reported. Here, we report a case of early infantile epileptic encephalopathy/Ohtahara syndrome (EIEE/OS) secondary to sporadic de novo heterogeneous mutations in SCN2A and GRIN2A genes in a phenotypically normal neonate with structurally normal brain who presented with intermittent, clusters of intractable seizures associated with ictal vocalizations. To our knowledge this is first such case report.
Case Presentation
A female baby of African American descent was born at 37 weeks and 5 days gestation age with a birth weight of 3118 g to a 35-year-old G7P6 mother via spontaneous vaginal delivery. Both parents were healthy and non-consanguineous. Pregnancy, labor and delivery were uncomplicated except for the fact that mother was group B streptococcus (GBS) positive. On day 2 she was noted to have lip smacking, eye rolling, and tremors and was transferred to the neonatal intensive care unit. She was given a loading dose of phenobarbital a complete septic workup was performed. Blood, urine, cerebrospinal fluid (CSF) cultures (bacterial and viral) were obtained. In addition, extensive metabolic workup was done and viral serological and polymerase chain reaction (PCR) tests were obtained. Empiric antibiotics and acyclovir were initiated. She continued to have multiple episodes of seizures despite escalation in anticonvulsant therapy, which included phenobarbital, fos-phenytoin, clonazepam, and levetiracetam. The seizures consisted of multiple episodes of itcal vocalizations associated with eye rolling, flailing of extremities, tachycardia, tachypnea, and desaturations. These episodes lasted for less than 1 minute and occurred in clusters multiple times per day. On clinical examination, the baby was nonsyndromic with no evidence of neurocutaneous markers but had one single right ear pit, poor Moro’s reflex, poor tone, no suck, and poor grasp. Electroencephalogram (EEG) showed bilateral frontal lobe epileptiform discharges, which transitioned to hypsarrhythmia followed by hypsarrhythmia with burst-suppression pattern. Magnetic resonance (MR) imaging, MR angiography, MR venography, and spectroscopy of the brain were normal. Echocardiogram, abdominal, pelvic and renal ultrasounds were normal. Therapeutic trials with thiamine, pyridoxine, folinic acid, and pyridoxal phosphate, and high-dose steroids demonstrated no benefit. Oxcarbazepine was added without improvement, and hence discontinued. Because of early onset of intractable, multidrug-resistant seizures, developmental delay, and EEG showing epileptiform discharges with burst suppression pattern epileptic encephalopathy syndromes were considered and 70 Gene Comprehensive Epilepsy Panel (CEP; which includes Sanger sequencing, array comparative genomic hybridization [CGH] and WES) and CSF neurotransmitter studies were obtained. Therapeutic trials with lacosamide, topiramate, and dextromethorphan were tried but failed. The seizures worsened with lacosamide, and thus it was discontinued. Results of the infectious and metabolic workup were negative and are shown in Tables 1 and 2, respectively. The CSF neurotransmitter studies were normal. The CEP revealed 2 de novo mutations: SCN2A c. 788C>T heterozygous mutation, p.Ala263Val and GRIN2A c.1354G>A heterozygous mutation, p. Val452Met. A diagnosis of EIEE/OS was confirmed and she was discharged home at age 2 months 21 days on phenobarbital, clonazepam, levetiracetam, and topiramate. At the time of discharge, the seizure frequency had decreased considerably.
Infectious Workup.
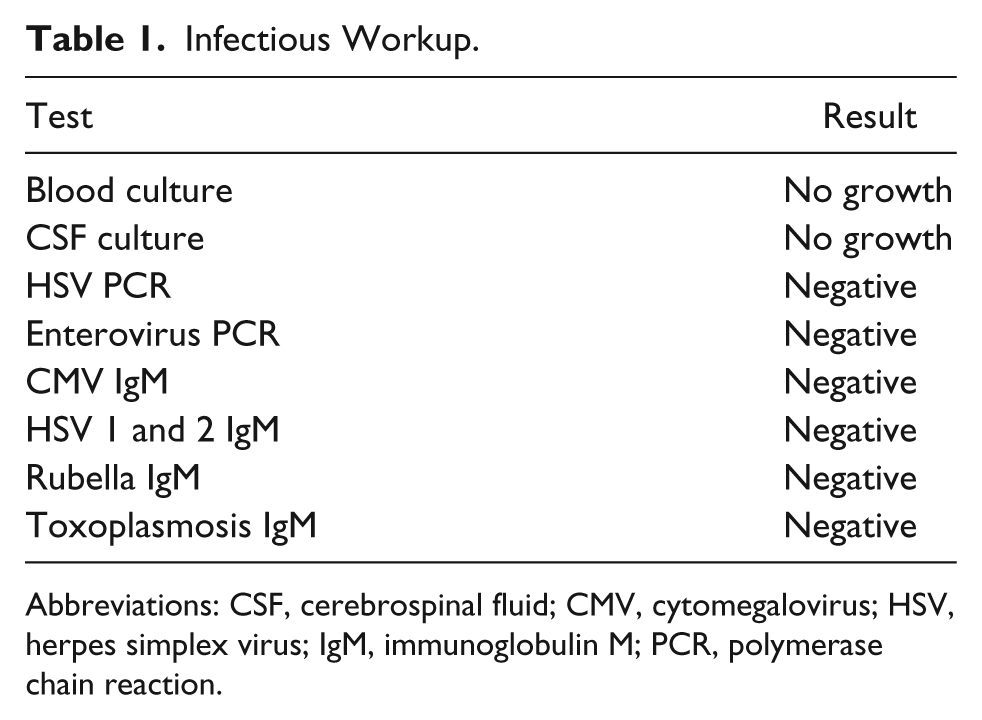
Abbreviations: CSF, cerebrospinal fluid; CMV, cytomegalovirus; HSV, herpes simplex virus; IgM, immunoglobulin M; PCR, polymerase chain reaction.
Metabolic Workup.
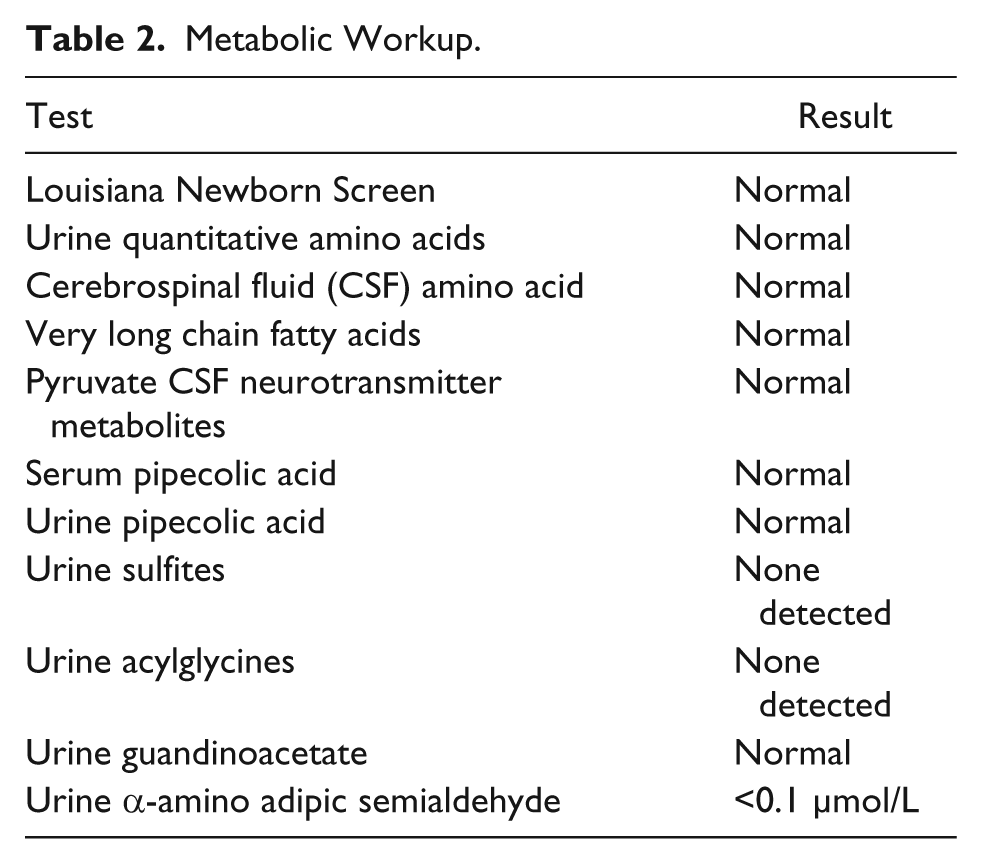
Discussion
Ion channels are specialized transmembrane proteins that facilitate passive flow of ions based on their electrochemical gradients. These ion channels (classified as sodium, potassium, calcium, or chloride ion channels) play important roles in generating and propagating action potentials, which result in cellular activities like neurotransmitter release and muscle contraction. These channels are activated by factors such as membrane potentials (voltage-gated), hormones, or neurotransmitters (ligand-gated). Generally, these ion channels are formed by multiple subunits like alpha or beta subunits, which are encoded by separate genes. 5 The advent of new techniques like WGS and WES has resulted in identification of various mutations in the genes encoding for these ion channels that have been classified as “channelopathies.” A large proportion of these channelopathies have been reported to be responsible for EIEE and other serious genetic disorders involving various organ systems. 5
Voltage-gated sodium channels consist of 1 α subunit and 1 or 2 auxiliary β subunits. Nine different isoforms of α subunit have been identified that includes Nav1.1–Nav1.9. SCN1A, SCN2A, SCN3A, and SCN8A genes encode the Nav1.1, Nav1.2, Nav1.3, and Nav1.6 isoforms, respectively. These isoforms are highly expressed in the human brain and mutations in the genes encoding these isoforms cause epilepsies and other disorders. 6
The excitatory neurons contain a predominance of Nav1.2 (SCN2A) and Nav1.6 (SCN8A) isoforms. The Nav1.2 isoforms are expressed early in development. During neuronal maturation these Nav1.2 isoforms (predominant in neonates and less excitable) are gradually replaced by Nav1.6 isoforms (predominant in adults and more excitable). The Nav1.1 isoforms (SCN1A) are predominantly expressed in neurons that release inhibitory neurotransmitter γ-aminobutyric acid (GABA). Hence, loss of function mutations in SCN1A gene leads to a reduction in Nav1.1 isoforms, thus increasing net excitability. Similar mutations have also been reported in SCN2A genes. SCN2A mutations that are inherited from an affected parent are usually benign but de novo SCN2A mutations affect excitatory neurons especially early during the developmental of neurons causing more severe early-onset epileptic encephalopathies.7-9
Similarly, N-methyl-
As described above, mutations occurring in SCN2A and GRIN2A genes have been described in literature but both mutations occurring in the same patient causing OS (as described in our case) has never been reported.
Ohtahara syndrome, first described in 1976 by Ohtahara, is a severe form of EIEE due to heterogeneous etiology. It is associated with variable EEG findings but the most specific and diagnostic EEG finding is the “suppression burst (SB) pattern,” which is characterized by periodic high voltage bursts of slow waves mixed with multifocal spikes, alternating with flat (isoelectric) lines occurring both during sleep and wakeful states with regular periodicity. This specific pattern may persist or transition into various other EEG patterns later in life. Although the data on exact incidence of OS is scant, it has been reported to occur in 1 in 100 000 live births. Seizures in children with OS are often refractory to multiple antiepileptic drugs and characterized most often by short duration (<10 seconds) tonic spasms. In about 30% of neonates, seizures occur within 10 days of life and in up to 70% of cases seizures manifest within 1 month of age. Various types of structural abnormalities of the brain like agyria, pachigyria, polymicrogyria, focal cortical dysplasia, megalencephaly, and others have been reported. 11 In our case, however, the patient had structurally normal brain and had severe multidrug-resistant seizures with ictal vocalizations. Ictal vocalizations occurred with every episode of seizure and this unique feature has also not been previously reported.
The pathophysiology of epileptic encephalopathies has been postulated to involve complex interaction between cortical and subcortical regions. Cortical outflow signals originating from a focal (eg, focal cortical dysplasia) or diffuse (eg, genetic mutations) region of abnormal irritable epileptogenic cortex results in altered cholinergic, serotonergic, and catecholaminergic function in subcortical regions resulting in diffuse cortical hypoactivity and encephalopathy. Interestingly, it is postulated that encephalopathy may not be the consequence of the seizures, but rather both of these may be products of the gene defect occurring independently. 12
Therapeutic strategies in the management of OS include both medical and surgical options, and the choice of treatment may be influenced by the etiology. Various antiepileptic medications like adrenocorticotropic hormone, oral steroids, clonazepam, chloral hydrate, levetiracetam, acetazolamide and zonisamide have been tried with some success in OS. In addition, high doses of vitamin B6, pyridoxal-5-phosphate, sodium valproate, phenytoin, topiramate, and vigabatrin have also been tried with poor results. 13 Ketogenic diet and in selected cases with known structural lesions in the brain surgical options like lobectomy, hemispherotomy, hemispherectomy or callosotomy has demonstrated promising results.14,15 The reason for worsening of seizures with lacosamide may be related to its novel mechanism of action affecting the slow rather than fast inactivation of the sodium channel. 16
Both the mortality rate and prognosis are very grave with poor quality of life and long-term outcomes. Genetic analyses are increasingly becoming important part of the diagnostic workup and helping the clinicians in choosing treatment tailored toward a specific channelopathy and perhaps improve long-term outcomes.
Conclusions
Recent improvements in genetic testing methods have greatly enhanced our diagnosing abilities of these rare mutations thus helping us establish genetic etiologies for the intractable seizures occurring early in life. This has facilitated discussions with families regarding risks of recurrence in future pregnancies. Future research about the structure and function of these ion channels will help in risk stratification and development of novel pharmacotherapies.
Author Contributions
DS, ML, TA, and YS conceptualized and designed the study, drafted the initial manuscript, reviewed and revised the manuscript, and approved the final manuscript as submitted. SN, LB, and OA carried out the initial analyses and approved the final manuscript as submitted. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.