
Abstract

Introduction
Chronic granulomatous disease (CGD) is a rare primary immune deficiency that is characterized by repeated infections with bacterial and fungal pathogens, as well as the formation of granulomas in tissue.1,2 It is a disorder of the NADPH oxidase system, culminating in an inability of the phagocyte to generate superoxide, leading to the defective killing of pathogenic organisms. We report the case of a 10-month-old male infant who presented with prolonged fever of unknown origin, persistent diarrhea, and failure to thrive. An extensive evaluation was nondiagnostic until CGD was suspected when a jejunal biopsy repeated for the third time showed the presence of a granuloma.
Case Report
A 10-month-old male infant presented with failure to thrive, daily fevers for 2.5 months, and diarrhea for 3 months. Multiple previous emergency room visits and one prior hospitalization had not yielded a diagnosis. He was born in the United States at 34 weeks gestation to nonconsanguineous parents from the Dominican Republic and was in the neonatal intensive care unit for 17 days for prematurity and feeding issues. Family history was notable for 3 half-siblings (2 males, 1 female, each by the same mother, but a different father) who all died within the first week of life. At 4 months of age, he had bloody diarrhea and was diagnosed with milk protein allergy, and his milk was changed to an extensively hydrolyzed formula. He tolerated this change well and was started on age-appropriate dairy-free foods around 6 months of age. He stopped gaining weight around this time and lost nearly 5 lbs by age 10 months. Developmental milestones were stagnant after 6 months. His immunizations were up to date.
On exam he was weak, tired appearing, and fussy. He had a sunken fontanelle and dry lips. The abdomen was distended and tympanic but nontender and distensible with no masses or hepatosplenomegaly. Cardiac, neurologic, and respiratory exams were normal. He was at the fifth percentile for weight and head circumference. Laboratory values were notable for anemia (hemoglobin 6.5 g/dL), thrombocytosis (platelets 858 000/µL), hypoalbuminemia (2.6 g/dL), and elevated C-reactive protein (12.8 mg/dL).
Infectious workup included multiple blood cultures, urine culture, Brucella serology, PPD (purified protein derivative), culture for AFB (acid-fast bacilli), HBsAg (hepatitis B surface antigen), hepatitis A IgG (immunoglobulin G) and IgM (immunoglobulin M), HIV ab/ag, EBV (Epstein-Barr virus), CMV (cytomegalovirus), adenoviral polymerase chain reaction, malarial smear, Lyme, bartonella serology, stool O&P (ova and parasite), culture and stool for cryptosporidium, all of which were negative. Immune evaluation showed elevated serum immunoglobulins (IgG 1840 mg/dL, IgA 226 mg/dL, IgM 121 mg/dL) and normal lymphocyte subsets. He had normal levels of FOXP3, which made immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) less likely.
Fecal calprotectin was elevated (>2000 µg/g, nL <152.9 µg/g) suggestive of inflammatory bowel disease. Computed tomography and magnetic resonance imaging of the abdomen and pelvis revealed scattered wall thickening in the small bowel and to a lesser extent the colon and rectum. Upper endoscopy and repeat colonoscopy showed focal villous blunting of the small intestines, and scattered pigment-laden macrophages and prominent paneth cells of the entire colon. He eventually required total parenteral nutrition for adequate caloric intake.
The patient continued to have daily fevers (Tmax 39.7°C). A positron emission tomography scan revealed multiple 18-fluorodeoxyglucose (FDG)-avid lymph nodes in the neck, mediastinum, abdomen, retroperitoneum, and bilateral inguinal regions and a FDG-avid lesion in the spleen concerning for lymphoma. Bone marrow biopsy was done and showed no evidence of non-Hodgkin’s lymphoma, and flow cytometry was not consistent with leukemia.
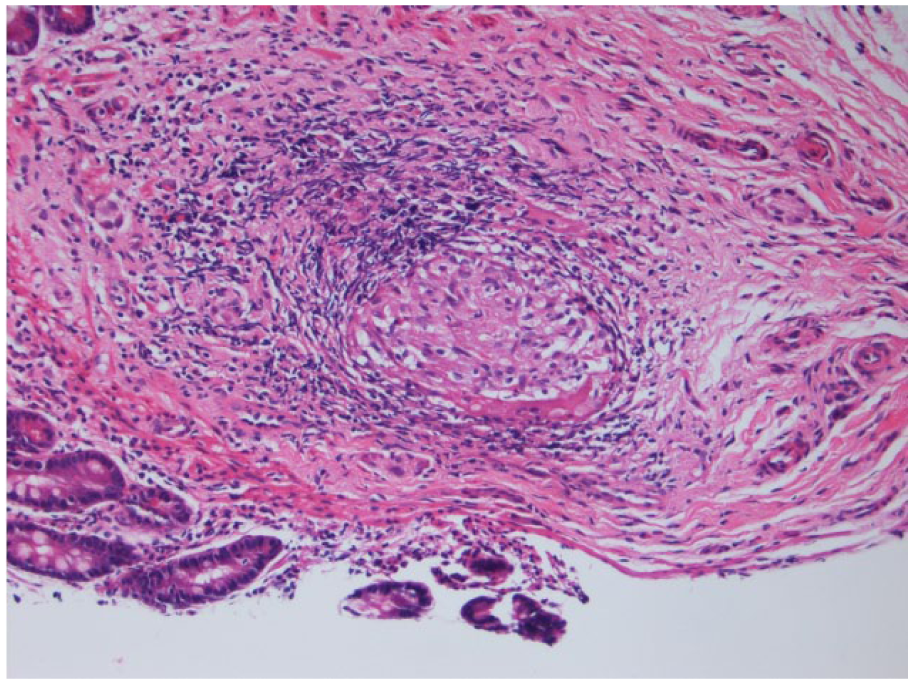
A repeat upper endoscopy was done with the jejunal biopsy showing small intestinal mucosa with active enteritis. Pathology of the tissue showed focal acute inflammation and a well-formed, nonnecrotizing granuloma (see Figures 1 and 1.1). Pigment-laden macrophages with fine, brown-yellow pigment were seen in the lamina propria of the colon (Figure 2).
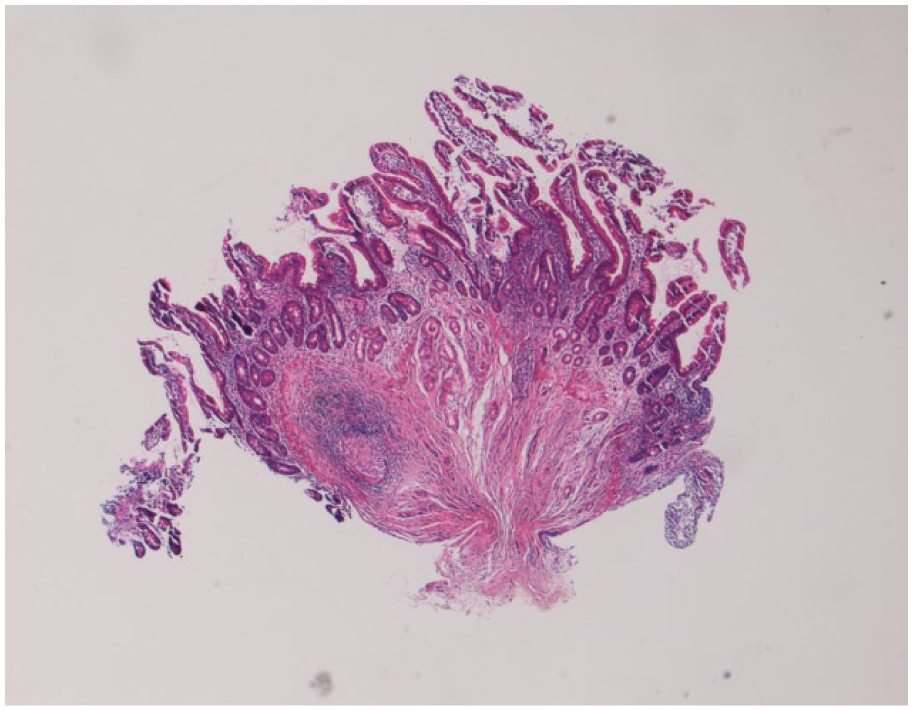
Jejunal biopsy from our patient containing a sharply defined, nonnecrotizing granuloma surrounded by lymphocytic inflammation (hematoxylin-eosin [H&E]) stain, magnification 4×. (1.1) shows the granuloma further magnified (H&E stain, magnification 20×).
Colonic biopsy from our patient containing pigment-laden macrophages within the lamina propria (hematoxylin and eosin stain, magnification 20×).
Based on the results from the biopsy, differential diagnosis was narrowed to sarcoidosis, early onset Crohn’s disease, and CGD. He was started on oral steroids and oxidative burst testing was simultaneously sent. Oxidative burst came back positive with neutrophil dihydrorhodamine (DHR) assay showing 0.4% of normal oxidase activity compared to control and this was confirmed on repeat testing.
CYBB gene sequencing of gp91phox of the NADPH oxidase complex revealed a R22BX nonsense mutation (c.676C>T, p.Arg226Ter variant) confirming the diagnosis of X-linked chronic granulomatous disease (XL-CGD).
An abdominal ultrasound done at this time showed hepatomegaly without focal lesions and splenomegaly with innumerable hypo-echoic avascular lesions, the largest being 1 cm, suggestive of multiple abscesses. He was empirically started on vancomycin, cefepime, and voriconazole to cover for bacterial and fungal abscesses, which can occur with CGD. He was also treated with corticosteroids for suspected CGD-associated colitis. Within a week of treatment, he became afebrile, diarrhea resolved, diet was advanced, and he started gaining weight. After 5 weeks of treatment his abdominal ultrasound revealed complete resolution of his splenic lesions. He is currently being treated with trimethoprim-sulfamethoxazole, voriconazole, prednisone, and interferon-γ while awaiting bone marrow transplantation.
Discussion
CGD represents a heterogeneous group of disorders characterized by defective generation of a respiratory burst in human phagocytes (neutrophils, mononuclear cells, macrophages, and eosinophils) and dysregulated inflammatory response. The inability to generate superoxide leads to an inability to contain certain infectious pathogens such as Staphylococcus, Nocardia, Aspergillus, Candida, atypical Mycobacteria, and gram-negative Enterobacteriaciae. 1
Most CGD patients present with infectious illness, which include sinopulmonary disease, abscesses, lymphadenitis, or osteomyelitis. Clinical symptoms and signs can also result due to inflammation or structural disease and resulting organ dysfunction. 1 The most commonly affected organs are the gastrointestinal (GI) tract (88.2%), lungs (26.4%), urogenital tract (17.6%), and eyes (8.8%) according to one cohort study. 3 Failure to thrive is also common, due to the nutritional effects of chronic infection and inflammation. 4
Recent studies highlight the high prevalence of gastrointestinal disease in patients with CGD, particularly XL-CGD. From a registry of 368 CGD patients, Winkelstein et al reported that GI manifestations preceded the diagnosis of CGD in up to 17% of patients. 5 In another study of 87 CGD patients with mild to severe clinical GI symptoms, 95% were found to have demonstrable disease on pathologic evaluation of specimens obtained from upper or lower endoscopies. The X-linked form of CGD may be associated with more severe GI involvement. A National Institutes of Health study by Marciano et al found GI involvement in 32.8% of patients (46 of 140), 89% of whom had X-linked inheritance. The median age at the time of initial GI manifestations was 5 years (range = 0.8-30 years); 70% of the affected patients presented with GI involvement in the first decade of life. Abdominal pain was the most frequent symptom (100%), and hypoalbuminemia was the most frequent sign (70%). They found no clear evidence for an infectious cause and suggested that GI involvement should be sought in patients who have CGD with abdominal pain, growth delay, or hypoalbuminemia. 6
The lesions of CGD are discontinuous and may involve any part of the GI tract from the mouth to the anus though the colon is most commonly affected. Grossly, lesions can be characterized by ulcers, abscesses, fistulae, strictures, and obstructive symptoms. 7 Inflammatory granulomatous colitis can also lead to obstructive disease, diarrhea, malabsorption, or other manifestations. 7 Hepatic involvement by Staphylococcus aureus and Pseudomonas cepacia can manifest as granuloma or abscess formation. 8
Microgranulomas, pigmented macrophages, and tissue eosinophilia are commonly encountered in the GI tract and not associated with acute inflammation. 9 The inflammatory pattern within the gastrointestinal tract of patients with CGD can be mistaken for that observed with Crohn’s disease. However, the granulomata of Crohn’s disease are typically poorly defined, while those of CGD are sharply defined and surrounded by a cuff of lymphocytes. 10 Additionally, pigment-laden macrophages with fine, brown-yellow pigment are commonly present in the lamina propria of colonic biopsies, which is believed to be a histologic hallmark of CGD (Figure 2). The pigment-laden macrophages may be quite numerous and cause GI obstruction by infiltrating the viscous wall. 2 Both of these features were observed in our patient.
The mainstays of therapy in CGD have been antimicrobial agents for primary prophylaxis against bacterial and fungal infections and immunomodulatory therapy such as interferon-γ. Corticosteroids are recommended for inflammatory complications such as colitis followed by a slow taper. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only known cure for CGD. Clinical trials show that HSCT restores normal immune function of phagocytes leading to immune recovery and better survival in CGD patients with stable remission. 11
It is important to recognize that CGD can present with GI manifestations without any evidence of infection. Is should be considered on the differential in any pediatric patient with recurrent fevers of unknown origin, failure to thrive, and GI involvement. GI biopsy may be helpful as there are certain histologic features that are characteristic such as hemosiderin-laden macrophages and granulomas. Diagnosis requires evaluation of the neutrophil oxidative burst followed by genetic analysis for specific gene mutations with XL-CGD (CYBB mutation) being the most common and most severe. Early diagnosis is imperative for early management with antimicrobials, interferon-γ, corticosteroids, and referral for HSCT, which is curative.
Author Contributions
SM contributed to conception and design, drafted the initial manuscript and conducted the literature review. JT contributed to the immunology discussion, and edited the document. PA contributed in drafting the manuscript and editing the document. BU and WB provided the images and contributed to the pathology discussion. DG critically reviewed the manuscript and provided supervision. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.