
Abstract
Chronic granulomatous disease (CGD) is an inherited autosomal recessive or X-Linked primitive immunodeficiency (PID), due to a defective nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex impairing anti-infectious and anti-inflammatory role of peripheral blood mononuclear cells. It is characterized by severe bacterial and fungal infections and by excessive inflammation leading to granulomatous complications. This work was made over a period of 34 years on 41 Tunisian patients suffering from CGD. Cumulative follow-up of patients was 2768.5 months, median 31 months. Survival was studied by survival curves according to Kaplan-Meier method. Lymphatic nodes, pulmonary and cutaneous infections predominate as revealing manifestations and as infectious events during patients’ monitoring. At study end 12 patients died mainly of invasive pulmonary aspergillosis and septicemia. Median age of death was 30 months. CGD remains compatible with a decent quality of life. Early diagnosis, anti-infectious prophylaxis, and initiation of adequate management, as soon as complication is perceived, promote pretty good evolution.
Introduction
Chronic granulomatous disease (CGD) is an inherited and genetically heterogenous immunodeficiency disorder resulting from defects of one of the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex in phagocytic cells. Mutations in at least 5 different genes involved in the assembly and activation of the NADPH oxidase can lead to CGD. 1 NADPH oxidase has membrane-associated (eg, gp91-phox and p22-phox) and cytosolic components (eg, p47-phox and p67-phox). The gp91-phox defect is inherited in an X Linked (XL) manner, whereas the p22-phox, p47-phox, and p67-phox defects are inherited in an autosomal recessive (AR) manner. The X-Linked form usually more severe, presents earlier, and has a higher mortality.2,3
CGD is characterized by gene defects that can affect one of the various genes: CYBB, CYBA, NCF1, NCF2, NCF3, encoding, respectively, for, gp91phox, p22phox, p47phox, p67phox, and p40phox. The mutation of the CYBB gene located on the X chromosome is responsible for the XL CGD, whereas mutations at the other genes are responsible for the AR forms of the disease. These gene mutations are usually responsible for the absence of the corresponding protein or the presence of a nonfunctional protein resulting in a defective NADPH oxidase complex.4,5 In addition to its anti-infectious role, NADPH oxidase has a crucial role in regulating and controlling inflammation. Thus, peripheral blood mononuclear cells (PBMCs) from CGD patients had impaired activation of Nrf2, a redox-sensitive transcriptional factor that induces oxidant scavenging pathways and functions to limit cellular injury and inflammation. 6
CGD is characterized by severe bacterial and fungal infections and excessive inflammation. In addition, this defect predisposes to granulomatous complications and autoimmune diseases.5,7
Although most CGD patients are diagnosed as toddlers, it can present in infancy and even in adulthood. However, a growing number of patients are diagnosed in later childhood or adulthood.2,8
The frequent sites of infection are lung mainly pneumonia, skin mainly abscesses, lymph nodes (lymphadenitis), osteomyelitis, and sepsis. 9 Perianal abscesses and gingivitis are also common.2,10 The microbiology of infections in CGD is remarkable for its relative specificity. The overwhelming majority of infections in CGD are due to only a limited number of organisms: Staphylococcus aureus, Burkholderia (Pseudomonas) cepacia complex, Klebsiella pnuemoniae, Salmonella species, Serratia marcescens, Nocardia species, and Aspergillus species.
In addition to recurrent infections, CGD is also characterized by abnormally exuberant inflammatory responses leading to granuloma formation, such as granulomatous enteritis resembling Crohn’s disease. 11 The urogenital tract is the second most commonly affected organ with genitourinary obstruction 12 followed by the lungs 13 and eyes. 14
Despite the significant progress made in antibiotic and antifungal therapy and prophylaxis, patients with CGD still develop serious infections. The US National Institutes of Health (NIH) has followed more than 250 patients with CGD over almost 40 years, the majority of whom were diagnosed following infections of skin, lymph node, lung or liver. The diagnosis was usually established early in the life (median age of diagnosis 5.4 years), although a small proportion were diagnosed as adults. Notably, the majority of these later diagnoses were due to AR forms of CGD.1,15
Diagnosis of CGD is based on the demonstration of an absent respiratory burst. 16 Diagnosis of CGD can be made by either by the Nitroblue Tetrazolium (NBT) dye reduction test 17 or using flow cytometry based Dihydrorhodamine (DHR) assay, which is a more objective test. 18 This test represents a very sensitive and specific assay that reliably detects all NADPH oxidase deficiencies in neutrophils.17,19
We tried in this article to emphasize majors’ clinical and paraclinical aspects of CGD. Prediagnosis and postdiagnosis particularities are put in light, as well as monitoring, treatment (prophylactic and cure), and the evolution of this disease. Data of patients’ future, survival, and quality of life conclude this work. Twelve patients died during the work period, principaly by invasive pulmonary aspergillosis and septicemia.
Patients and Methods
Patients
This is a retrospective epidemiological study led over 34 years (from January 1983 to December 2017) of 41 patients with CGD at the Bone Marrow Transplant Center (Tunis, Tunisia).
Methods
Biological diagnosis
CGD diagnosis was established for earlier and later patients, respectively, by Nitroblue Tetrazolium test (NBT) (37 patients) and flow cytometry-based Dihydrorhodamine (DHR) 123 assay (4 patients).
Innate and adaptative immunity were analyzed, respectively, by immunoglobulin assay, B (CD 19, CD20) and T cells immunophenotyping (CD3, CD4, CD8). Lymphocytes functionality was checked by lymphocyte proliferation tests.
Aspergillus infections were defined either by galactomannan positive assays and/or by other clinical and paraclinical characters.
The inheritance mode of the disease was evaluated by a family tree study, since we not dispose of genetic study.
Follow-up
Clinical, biological, and follow-up informations were collected on a datasheet. The infectious episodes considered correspond to cases where an organism has been identified during clinical manifestations and / or in cases where systemic antibiotic therapy has been necessary. All inflammatory complications and also therapeutics administrated to patients were reported. All patients have had prophylactic antibiotic (Co-trimoxazole [25 mg/kg/day]) and anti-fungus (Itraconazole [10 mg/kg/day]) therapy at CGD diagnosis. These therapies are reevaluated every 3 months. All patients were submitted every 3 months and a battery of clinical and paraclinical exams. They have had a chest radiography every 3 months looking for pulmonary aspergillosis and an abdominal echography, which is now more commonly used for diagnosis of deep abssess, particularly liver absess.
Statistical analysis
SPSS software (Statistical Product and Services Solutions, version 16.0, SPSS Inc, Chicago, IL, USA) was used for statistical analyses. Survival data were studied by establishing survival curves according to Kaplan–Meier method. Survival forecasts factors search was made in univariate analysis by comparing the survival curves by the Log Rank test.
Results
Epidemiological Characteristics of Patients
Among the 41 studied patients, 28 (68%) were male and 13 (32%) female. All 41 patients belong to 33 families, and parental consanguinity was found in 68% of patients (Figure 1). Seven patients (17%) had family histories of GCD and 24 (58%) of the studied families have a history of 43 young deaths in a primary immunodeficiency context.
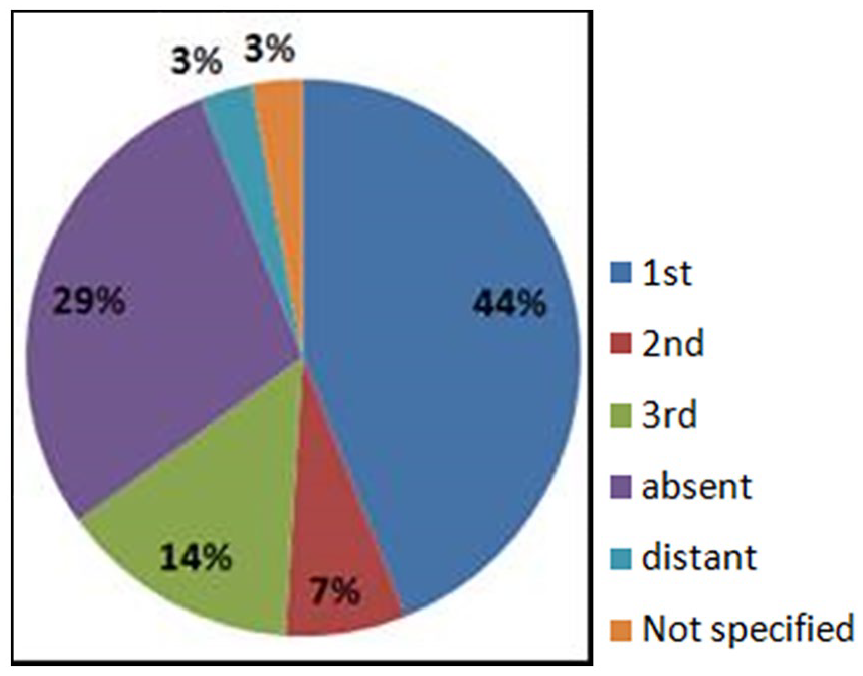
Patients repartition according to the degree of consanguinity.
Clinical Study
Thirty six (88%) of patients developed the most frequent clinical symptoms commonly observed during this pathology. Lymphatic nodes proliferation, pulmonary, and cutaneous infections were the predominant types of symptoms present in our patients. In 75% of cases, those symptoms developed during the first year of life, 17% between 1 and 5 years, 5% between 5 and 10 years, and only 1 patient developed symptoms after the age of 10 years. Thirty one patients had BCG vaccine at birth and 17 among them developed BCG-itis, which was respectively, generalized 6 (35%), locoregional 6 (35%), and local 5 (30%).
CGD diagnosis was made at age 19 months in median (range: 1-683 months). For 16 patients (39%), CGD diagnosis was made before age 1 year, and then for 18 patients (44%), 3 (7%) and 4 (10%) patients, respectively, between ages 1 and 5 years, 5 and 10 years, and over 10 years. One of the patients with a liver abscess was 57 years old at CGD diagnosis. This patient had a Klebsiella pneumoniae septicemia, associated with 5 liver abscesses, a purulent casting of the right eye and severe hyalite of the left eye, responsible for a sequellar cecity.
Time limit between clinical symptoms onset and CGD diagnosis was 10 months in median (range: 0-135 months). For 2 patients born before 1985, CGD was made after 8 years and over 2 years for 9 patients (22%). This time limit was of 10 months for both patients born, respectively, after (range: 0-39) and before (range: 0-135) year 2000, no statistically significant difference was found between these 2 groups.
Eleven patients (27%) had a delayed height-weight development and only 6 (15%) a delayed weight development. Also 11 patients had hepatosplenomegaly, and 4 (10%) had an isolated hepatomegaly or splenomegaly.
Paraclinical Findings
Complete blood count
Anemia was found in 29 patients (70.7%), it was microcytic and normocytic, respectively, in 83% and 13% of cases. Twenty patients (49%) had hyperleucocytosis, and 20 patients (48.7%) had thrombocytosis.
NBT and DHR123 assays
All patients have had a semiquantitative NBT assay, for 30 of them (73%), NBT reduction was complete and weak for the 27 % remaining. DHR123 assay was performed for only 4 patients, and values were, respectively, 0%, 1%, 10%, and 10%.
Immunoglobulin assay
Respectively, 22 (53%) and 20 (49%) of patients had elevated IgG and IgA values.
Lymphocytes subpopulation count
Lymphocytes subpopulation count was performed for only 54% of patients. T CD8+ elevated count was found for 12 patients (34%), and T CD4+/ TCD8+ ratio was elevated for 5 patients (12%).
Lymphocyte proliferation tests
Lymphocyte proliferation tests were performed entirely for 37 patients (90%), results showed a normal proliferation ranging, respectively, from 24% for Candida, 32% Tetanos, 46 % anti-CD3, 49% PHA, and 51% Tetanus.
Genetic study
As we had no access to genetic studies, genetic inheritance mode study was based on family tree study, which had revealed that for 27 patients (66%), CGD inheritance mode was AR, and that it was X linked for 12 patients (29%). For 2 patients (5%), we could not determine the inheritance mode.
Treatment and Patient Monitoring
Cumulative follow-up of patients was 2768.5 months, median of 31 months (range: 0-336).
Prophylactic treatment
Twenty eight patients (68%) have had Co-trimoxazole and Itraconazole at CGD diagnosis, 3 patients (7%) were lost from view after CGD diagnosis, 2 patients (5%) received first ketoconazole treatment, and 3 died soon after CGD diagnosis. Those treatments were well tolerated by patients. Five patients (12%) received voriconazole as antifungal and 9 (22%) INF-γ following severe infections.
Infectious episodes
One hundred twenty-seven infectious episodes were identified over the monitoring period, what means 0.045 episode/patient month (Table 1). Among those infectious episodes, 40 severe infections (31.5%) requiring hospitalization and intravenous antibiotic therapy were identified, what means 0.014 severe infection/patient month.
Percentage of Different Infectious Episodes During Patient Monitoring.
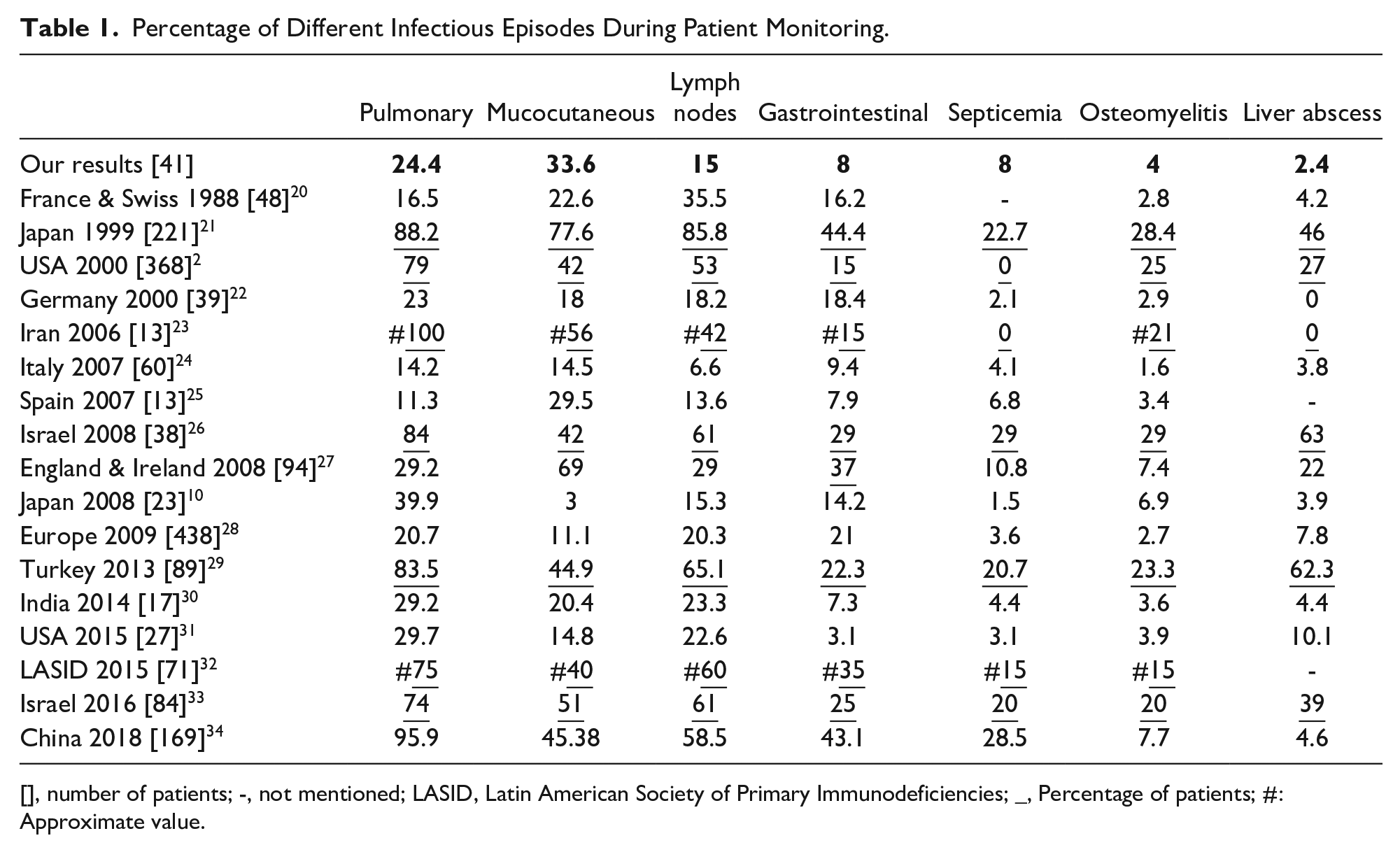
[], number of patients; -, not mentioned; LASID, Latin American Society of Primary Immunodeficiencies; _, Percentage of patients; #: Approximate value.
Among infection episodes, cutaneous infections predominated with 43 episodes (33.6%), followed by pulmonary infections (24.4%) and lymphatic nodes infections (15%) (Figure 2).
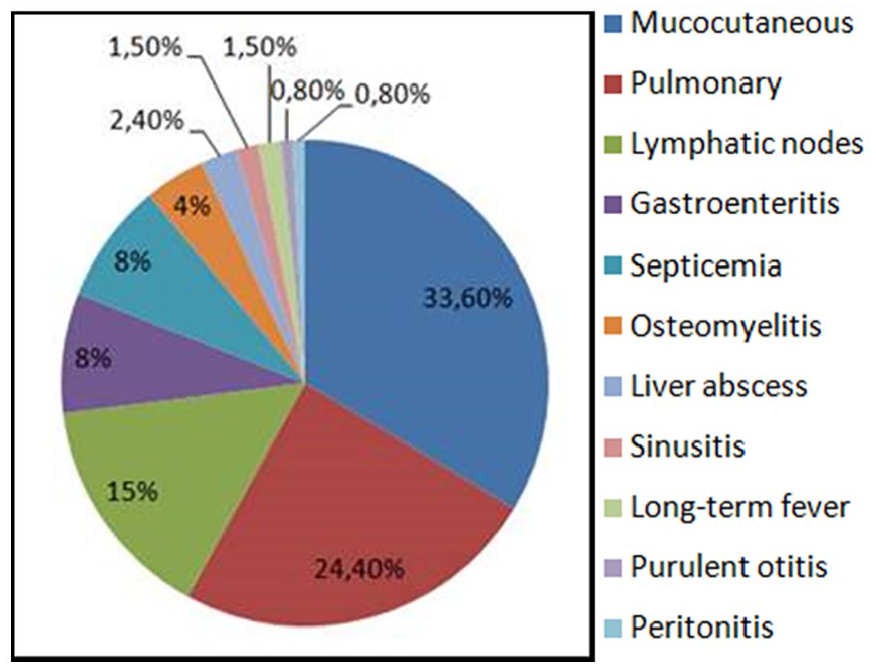
Infectious events during patient monitoring.
Microbiological profile
Microbiological study concerned 102 infectious episodes, representing 80.3% of all infections (Table 2). Microorganisms have been found only in 75.5% of explored episodes. Among those microorganisms, 18 types of organisms were identified in 86 samples. Bacteria was identified in 58 samples (67%), fungus in 18 (21%), parasites in 5 (6%) and viruses in 5 (6%) samples.
Percentage of Germs Commonly Found in Infectious Complications During CGD.
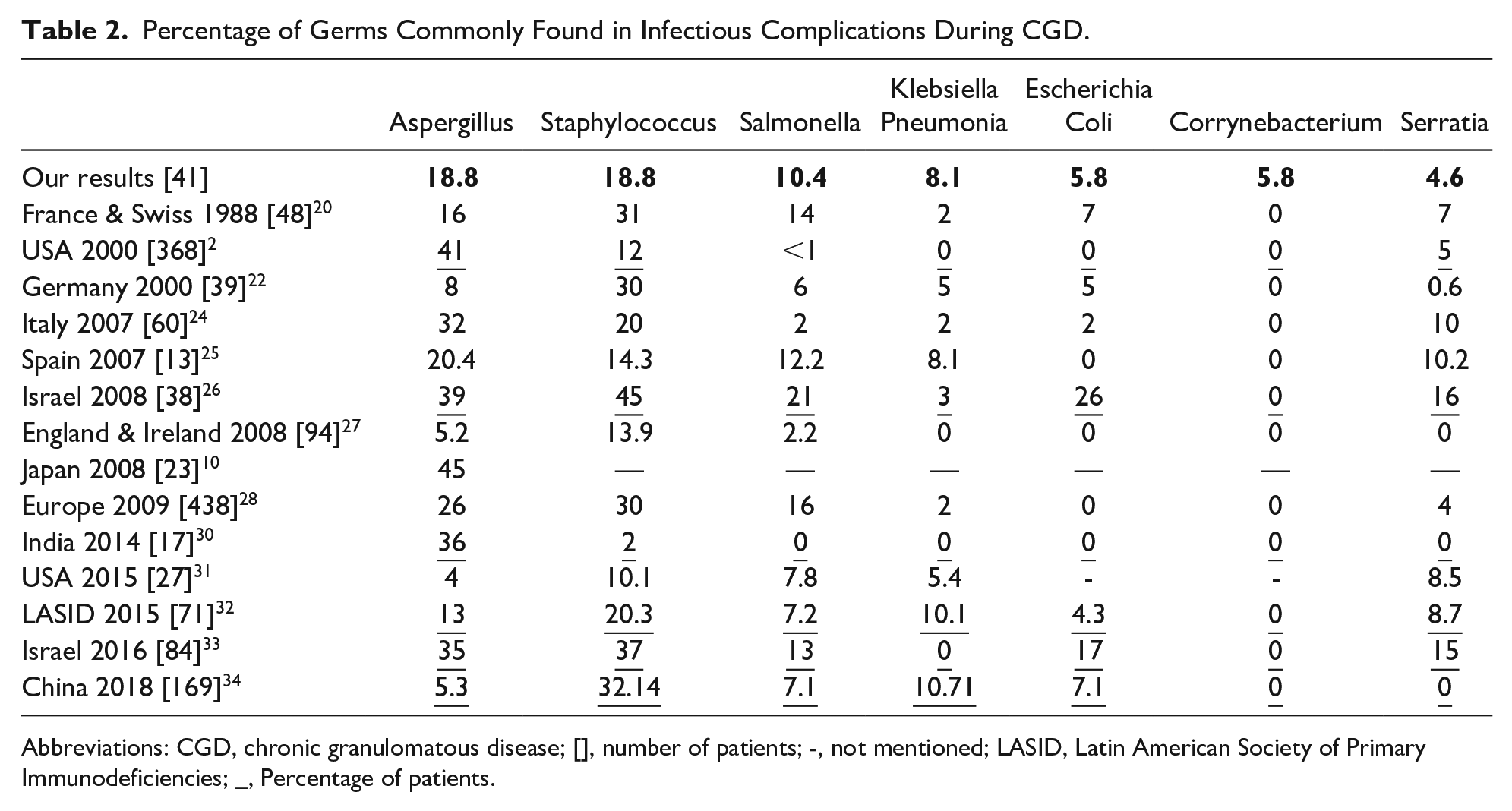
Abbreviations: CGD, chronic granulomatous disease; [], number of patients; -, not mentioned; LASID, Latin American Society of Primary Immunodeficiencies; _, Percentage of patients.
Mainly isolated organisms were Staphylococcus and Aspergillus each in 16 infectious episodes. Galactomannan tests were positive, negative, and not made, respectively, in 31%, 50% and 19% of cases. For those positive the distribution was as follow, 13 pulmonary invasive infections, 1 osteomyelitis and 2 liver abscesses.
Management and evolution of infectious episodes
Forty-three mucocutaneous infections of different types and localizations were observed for 18 patients with an average of 2.4 infections/patient, and were distributed as follow, cutaneous abscess (26%), oral trush (21%), perianal abscess (16%), and miscellaneous infections (pustular dermatitis, boil, paronychia, etc) (37%). Thirty nine of these infections were explored, and organisms were found in 28 cases.
Thirty one pulmonary infection episodes were reported, which represented 24.4% of all identified infections, only 27 of them were explored. So we have identified 13 aspergillosis, 4 Haemophilus influenzae infections, 1 Staphylococcus infection and no organisms were identified for the 9 remaining infections. Ten patients have had pulmonary aspergillosis at a mean age of 67.5 months (range: 23-132). Note that for 2 patients, such infections were CGD revealing. Evolution after treatment (amphotericin B, voriconazole, flucytosine) was favorable for 9 patients, but unfortunally 1 patient had lost sight. Infection recurrence affected 3 patients, 1 of which died later of skin fistulization with costal involvement.
Nineteen lymph node infections were diagnosed, 14 infectious nontuberculous adenitis, 4 adeno phlegmons and 1 lymph node tuberculosis. Organisms commonly found were found in 17 pulmonary infections, respectively, 13 with Aspergillus and 4 with H influenzae; 16 Staphylococcus infections (10 mucocutaneous, 4 of lymph nodes and 1 pulmonary); 6 with K pneumoniae (3 mucocutaneous and 3 of lymph nodes), 5 Cytrobacterium infections (3 mucocutaneous and 2 of the lymph nodes). Other organisms were less fond, as Esherichia Coli, Enterobacter, Serratia, Proteus, Mycobacterium tuberculosis, Candida albicans, and so on.
Among the 10 gastroenteritis infectious episodes diagnosed in 7 patients, 8 were explored. Organisms implicated were, Entamoeba histolitica (4 cases), Salmonella (2 cases), and in 1 case, respectively, Giardia and Rotavirus. Evolution was favorable except for 1 patient who developed a Salmonella septicemia.
Ten episodes of septicemia occurred in 10 patients; respectively, 7 were due to Salmonella typhi, 1 to E coli, 1 to Staphylococcus, and 1 to K pneumonia. Four among these patients died of septicemia, respectively, 2 to Salmonella, 1 to E coli, and 1 to K pneumoniae. Five patients experienced each one an osteomyelitis episode, due, respectively, to Serratia (2 cases), C albicans (1 case), Aspergillus (1 case) and no organisms was identified in 1 case. Mean age onset of these infections was 72.2 months (range: 14-132). Osteomyelitis reached vertebra, hand and feet bones, frontal bone, long bones and mastoid bone. Evolution was favorable for 3 patients, 1 developed a chronic osteomyelitis and 1 had a C albicans cerebral abscess secondary to mastoid bone lesion which resolved gradually weeks later after surgical drainage, Amphotericin B and fluconazole medical treatment. Note that additionally to medical treatment, 3 patients had interferon-gamma therapy.
Other infectious episodes occurred for some patients, as liver abscess (3 patients), organisms implicated were Aspergillus, Proteus and Enterobacter; sinusitis (2 patients) due to H influenza, and peritonitis (1 patient), acute media otitis (1 patient) and Long-term fever (2 patients).
Inflammatory and granulomatous episodes
Ten patients had hepatomegaly and 12 splenomegaly and 1 patient had a scrotal granuloma . One patient had a Crohn-like colitis which became corticosteroid dependent, only TNFα has allowed a favorable evolution after months of cellcept and IFN-γ therapy. Six patients have had atopic eczema lesions.
Patients’ Future, Survival and Quality of Life
At December 31th 2017, 22 patients (54%) were alive, 12 (29%) died and 7 (17%) were out of sight. Invasive pulmonary aspergillosis and septicemia were the most common death causes, (Table 3). Median age of death was 30 months (range: 7-253).
Major causes of Death by Number of Deceased Patients.
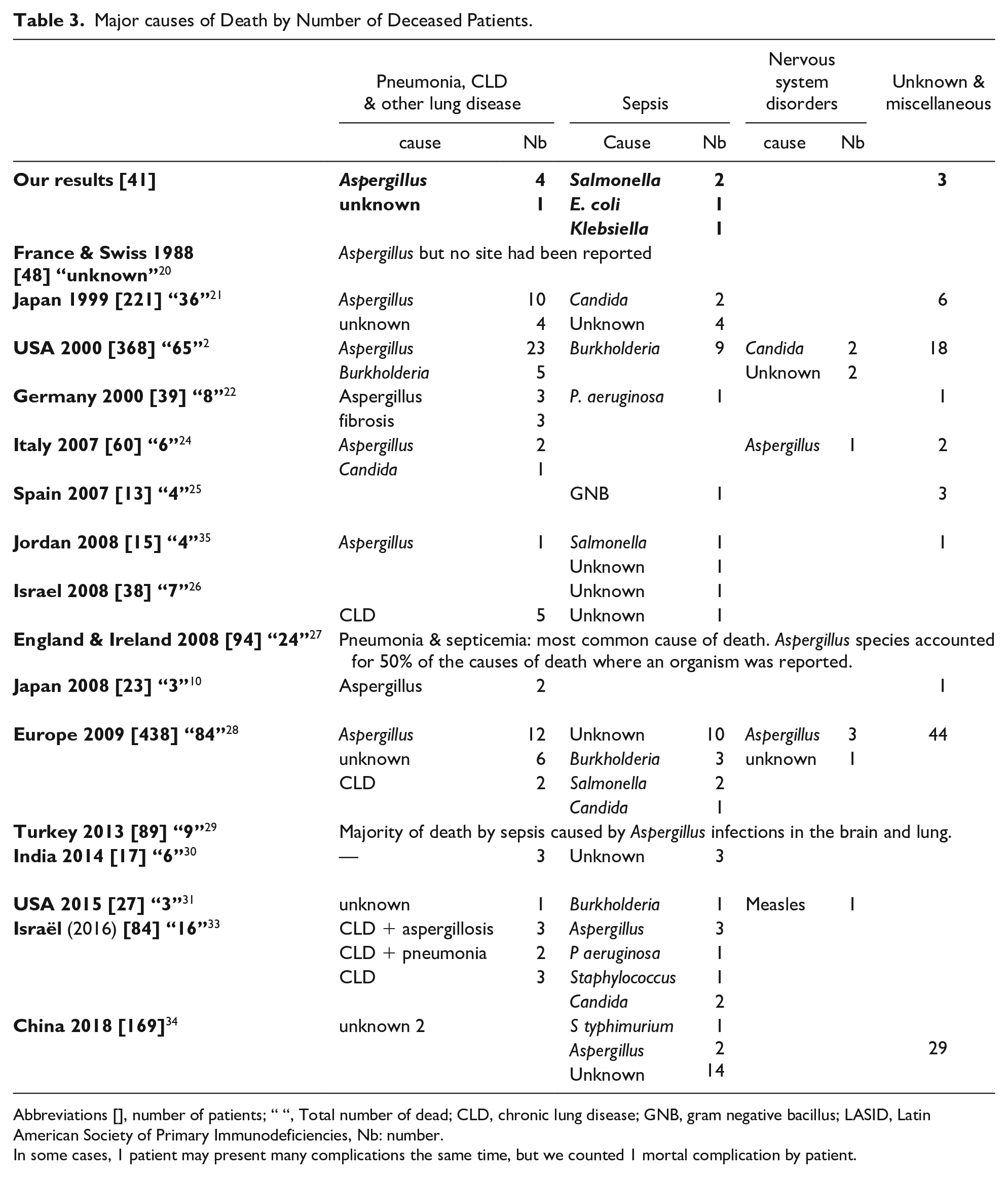
Abbreviations [], number of patients; “ “, Total number of dead; CLD, chronic lung disease; GNB, gram negative bacillus; LASID, Latin American Society of Primary Immunodeficiencies, Nb: number.
In some cases, 1 patient may present many complications the same time, but we counted 1 mortal complication by patient.
Overall survival
The average survival rate and the average death rate were respectively, 53.6% and 29.2% (Table 4). The overall survival during the first year of disease was 74% and decreased to 71% after 5 years of evolution and persisted until 20 years of evolution. After 20 years and until 30 years of disease evolution, overall survival persisted constant at 47% (Figure 3).
Survival and Mortality Rates in CGD in Some International Studies.
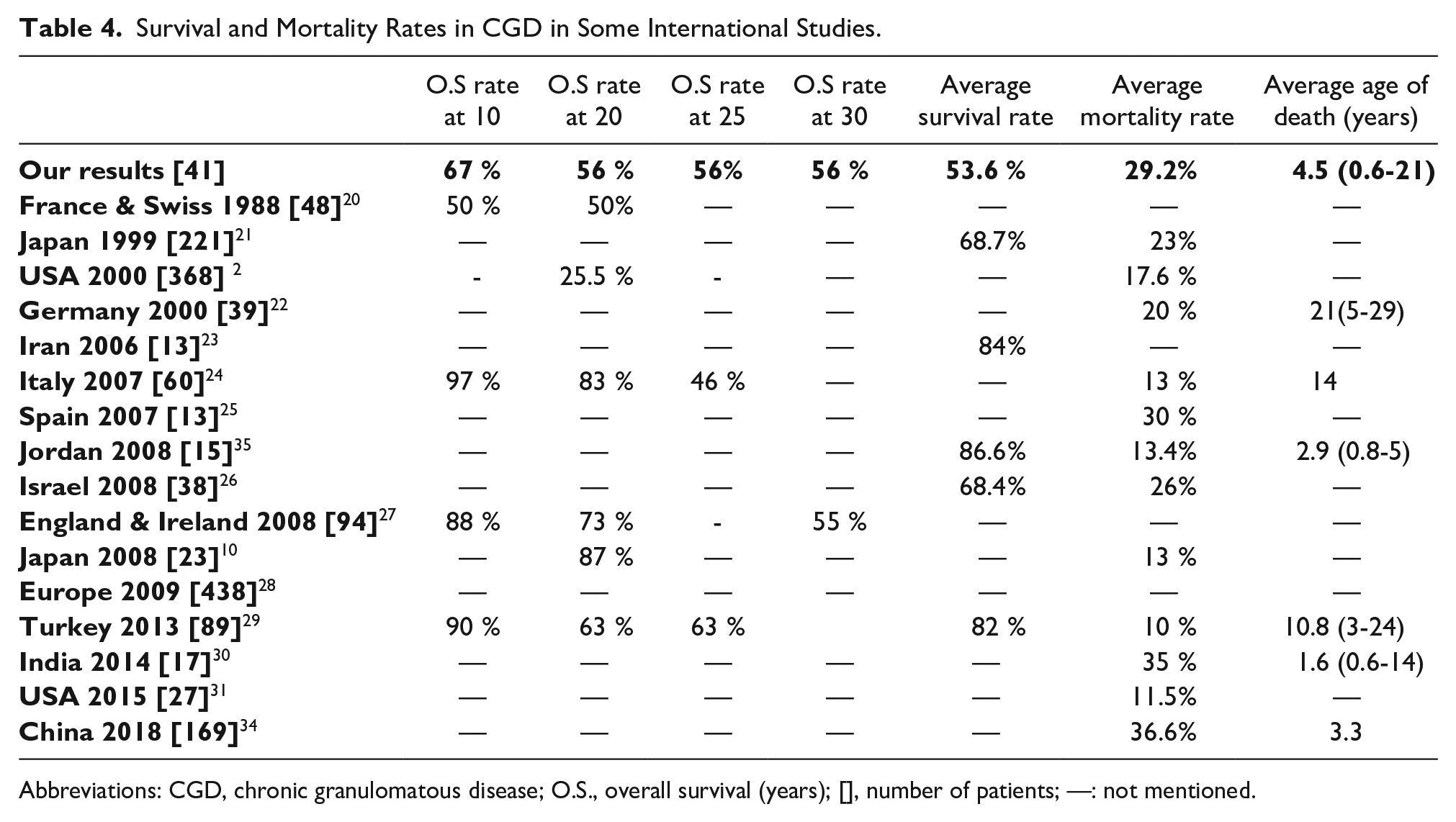
Abbreviations: CGD, chronic granulomatous disease; O.S., overall survival (years); [], number of patients; —: not mentioned.
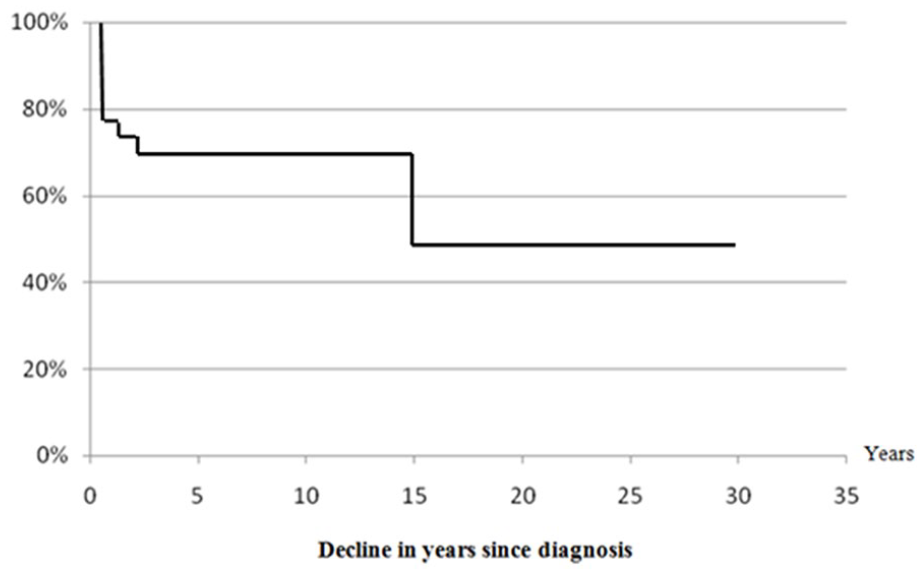
Overall survival curve according to monitoring years.
According to age, overall survival of patients without considering the date of diagnosis was 92% at 1 year of age and 72% at 2 years of age. Between ages 5 years to 20 years, overall survival was 67% and between ages 20 years to 30 years it decreased to 56% (Figure 4).
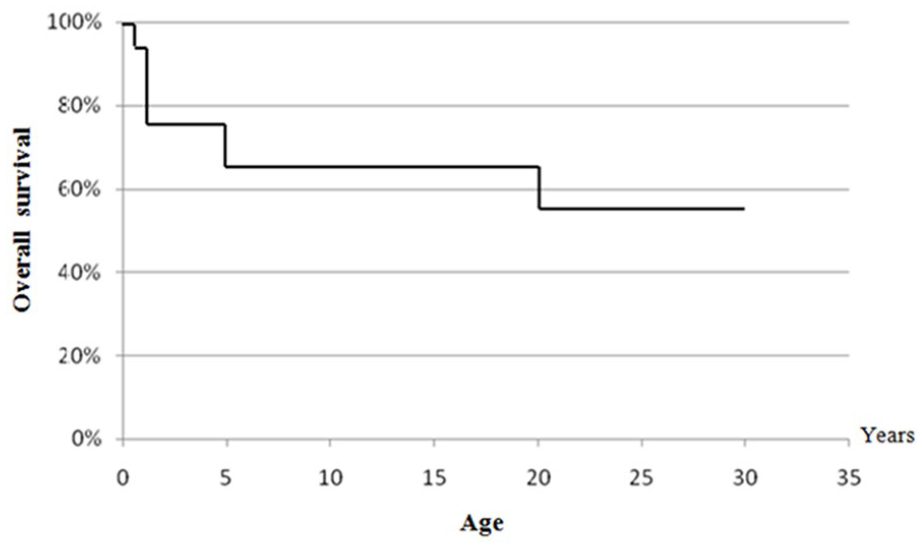
Overall survival curve as a function of age.
According to the genetic likely inheritance mode, overall survival with respect to age in the AR forms of the disease seems to be higher than this in forms linked to X, but there was not a statistically significant difference (Figure 5 and Table 5).
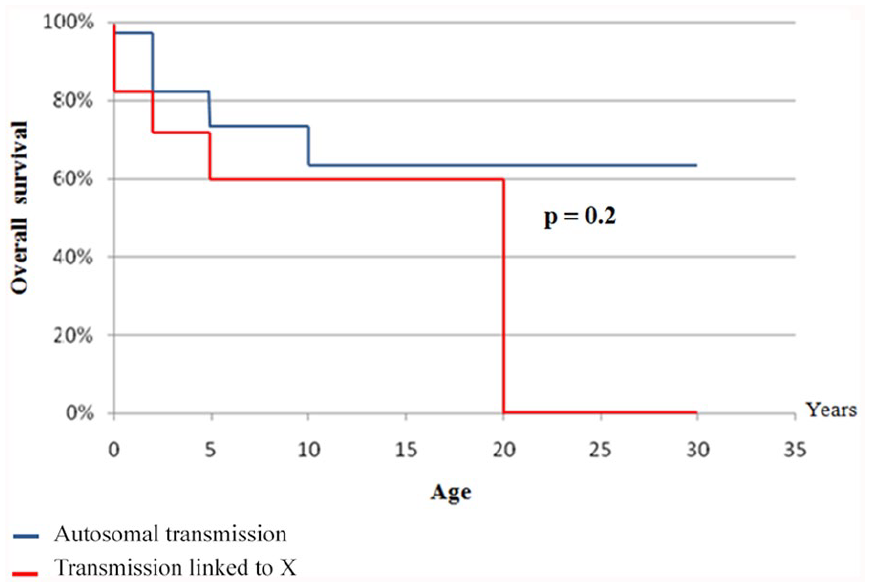
Global survival curve in relation to age according to the disease inheritance mode.
Survival and Mortality Rates in CGD in International Studies According to Its Genetic Transmission Modes.
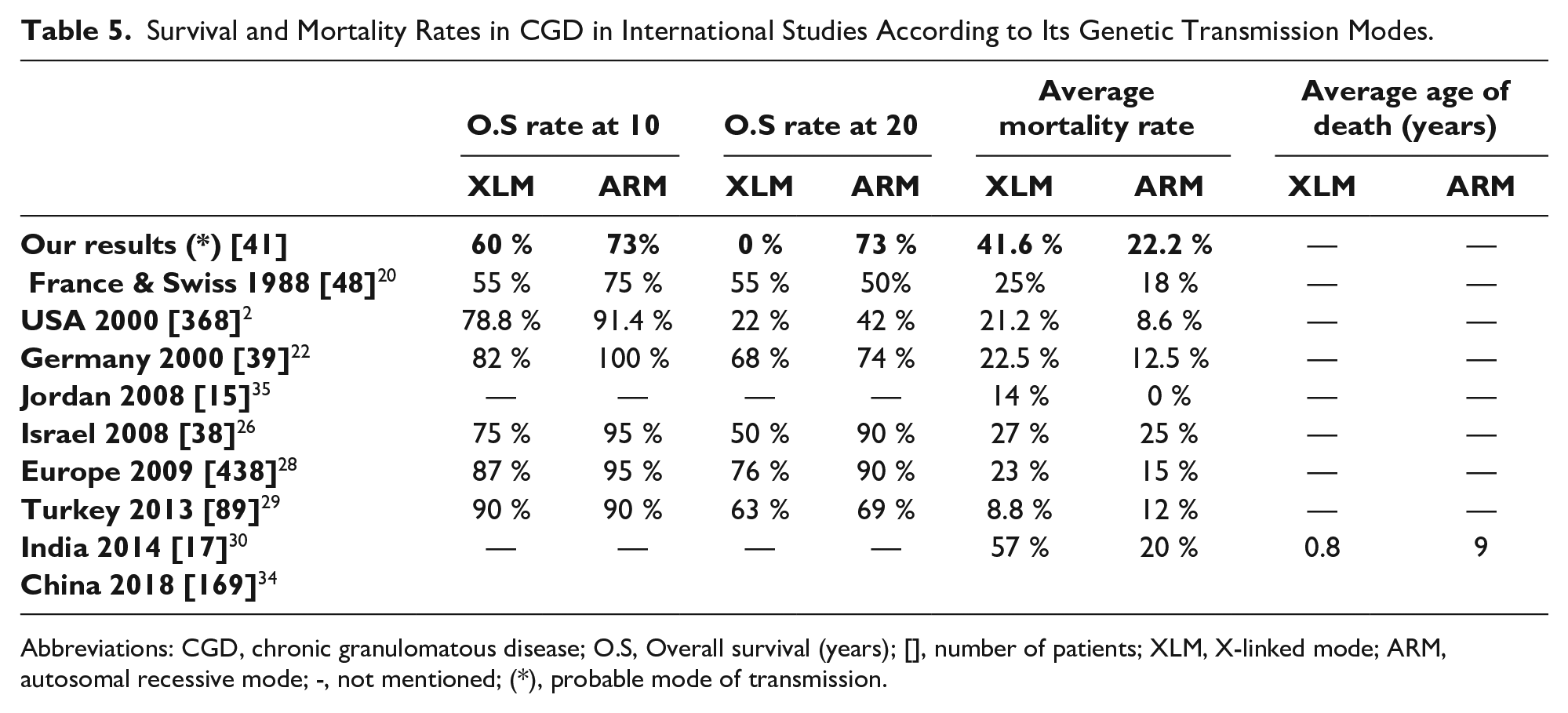
Abbreviations: CGD, chronic granulomatous disease; O.S, Overall survival (years); [], number of patients; XLM, X-linked mode; ARM, autosomal recessive mode; -, not mentioned; (*), probable mode of transmission.
Overall survival of patients treated with IFN-γ
The comparison of overall survival with a 10 years follow-up since CGD diagnosis, between the 9 patients that have received IFN-γ and those that do not, found no statistically significant difference between those 2 groups (Figure 6).
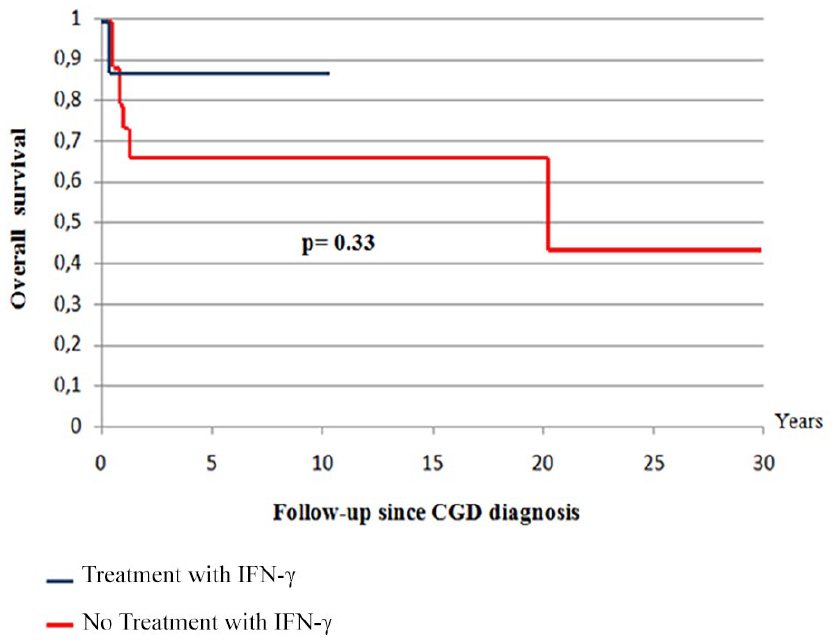
Overall survival according to IFN-γ treatment.
Quality of life
Twenty two patients were alive at the time of the study, among them, 13 have had a Karnofsky score higher than 80%, 7 a score of 70%, and 2 a score of 50%. The 2 oldest patients were born in 1983, so they were 34 years old at the time of the study. No patient experienced malignancies or hemopathies during monitoring.
Discussion
Survival According to CGD Duration Time
Overall survival was 74% in the first year of follow-up, from 5 until 20 years it stabilized at 71% and then between 20 and 30 years it decreased to stabilize at 47% (Figure 3). The relative stability of the survival rate after 5 years of follow-up may be explained by a better knowledge of the disease and its complications by physicians and a better management once CGD diagnosis has been established. Regular and rapid patients’ follow-up and specialized management of any complication, anti-infectious prophylaxis established as soon as CGD diagnosis is made, count also in survival rate stabilization. Whereas the fall of this rate to 47% during the third decade can be explained by occurrence of severe infections despite adequate management, due to the development of drug resistance. Overall survivals at 10 and 20 years were higher than those found in the study of Mouy et al 20 carried out in 1988, which would probably be due to the absence of effective antifungal treatment at that time. Those rates were below those found in many European studies carried out beyond year 2000, at, respectively, 10 years,24,27,29 20 years10,24,27,29 and 25 years,24,29 with the exception of the English serie 27 which shows at 30 years a slightly lower overall survival than ours.
Survival in Relation to Age
Since overall survival at 2 years of age was 72%, mortality in the first 2 years of life may be explained by the ignorance of primary immunodeficiencies (PID) by doctors who ensure the initial management of patients, the existence of children born before 1985 (where the disease was not well known and the care still not well codified), the epidemiological peculiarities of each country and also, existence of severe forms leading to rapid death (Figure 4). The plateau survival rate at 67% between 5 and 20 years can be explained by a lower susceptibility to infections at this age du to maturation of immune system. A lower rate of exposure to infections at these ages, as well as careful monitoring that contributes to better survival.
The average mortality rate in our study is close to that found in the Spanish series, 25 but it is higher than that found in many studies carried out in developed countries2,10,21,22,24,29,31,36 and some Middle East countries.23,26,35 Only a recent Indian study 30 showed a mortality average rate higher than ours. Whereas, the average survival rates were higher than ours21,23,26,29,35 (Table 4). Concerning average age of death, our results were lower than those of some European countries22,24,29 but higher than those of Jordan 35 and India. 30 The high rate of mortality in the first year of follow-up can be explained by: existence of severe forms of CGD leading to rapid death, difficulties in the management and medical resuscitation of these patients, erroneous diagnosis in cases where the clinical presentation has been modified by previous antibiotic therapy that was unjustified because abusive or not adapted to common CGD organisms.
Influence of CGD Inheritance Mode on Age Specific Survival and Disease Severity
The overall age-related survival was higher in likely AR forms that in probably XL forms (Figure 5). The absence of a statistically significant difference between the overall survivals of the 2 modes of inheritance of the disease is not very informative since there is no genetic study confirming the true mode of inheritance.
In our study, the overall 10-year survival rates for both XL and AR CGD were similar to those of the Mouy et al. 20 Whereas for survival at 20 years the differences between our results were clear, especially regarding the XL form. According to the different studies2,20,22,26,28-31,34,35 and even ours, survival rates at 10 and 20 years are higher for AR forms than for XL ones (Table 5). Köker et al 37 reported that the severity of the disease was related to the residual capacity of oxygen reactive forms and a residual activity of NADPH oxidase, which was thus associated with better survival.
Antecedents and Epidemiological Characteristics of CGD
In our study, CGD affects more boys than females, with a sex-ration of 2.15. In different studies2,10,20,26,28,30,31,34 there is also a male predominance. Parental consanguinity is found in 68% of the cases (Figure 1), which may explain the relative high frequency of AR forms. The European experience 28 noted a prevalence of these forms among immigrant groups from Arab countries and Turkey, which reflects the high frequency of consanguineous marriages in these communities. In our study, a history of confirmed CGD in the family was found in 17% of patients and allowed for earlier diagnosis of the disease as reported. 38 The recurrence of infant deaths also represent an indicator of PID.39,40 In our series, notion of infant death is found in 58% of families, with an average of 2.3 deaths per family, suggesting the existence of CGD familial forms of particularly severe expression.
Clinical Characteristics of CGD
Fifty-five percent of our patients developed a BCG-itis, which represented the most frequent complication of vaccination, it was nonlocalized in 65% of the cases. The frequency of these complications varies from one study to another, while for Fattahi et al, 38 60% of patients had BCG-itis, this frequency was 20% for Köker et al, 37 30% for de Oliveira-Junior et al 32 and dropped to 8% in the European experience. 28 Whereas, Zhou et al 34 reported, respectively, 46.2% and 13.1% of probable and definitive BCG infection. These differences may be explained by the fact that in Europe, except for France, BCG vaccination is not systematic. Some studies41-43 suggest a particular vulnerability of patients with CGD to BCG vaccine, hence the need to evoke PID and in particular CGD in the case of nonlocalized or trailing BCG-itis. Nonvaccination with BCG as a preventive measure for patients with a familial history of CGD prevents this complication, which can become fatal in its generalized form. It was thus recommended to contraindicate the BCG vaccine in CGD.31,41 Kawashima et al, 44 have suggested banning such vaccination before the age of 3 months. The production of tuberculosis vaccines from DNA fragments or ribosomal subunits could solve this problem.45,46
For our patients, infections of Lymph nodes, lungs and skin predominated as manifestations revealing CGD. While the main revealing manifestations of CGD are the same, their percentages differ from one study to another.2,13,21,34 While for some authors29,34,38 it was pneumonia, for others22,27 adenitis, cutaneous abscesses and pneumonia predominated. Recurrent pneumonia, pulmonary abscesses and adenitis constituted the bulk of CGD revelatory manifestations for some other studies2,24,26,30,32 For Bortoletto et al, 31 lymphadenitis and skin abscess predominated.
Cutaneous granulomas is the clinical forms of cutaneous involvement most suggestive of CGD diagnosis, thus they may localisate in different sites according to the initial infectious focus. In our study a particular localization was found for 1 patient, it was a voluminous suppurative nasal granuloma, responsible for a loss of substance of nasal cartilage. Such types of granuloma are reported to be one of a particular complications revealing CGD; it’s generally secondary to a long lasting infection.31,32,47
For our patients, three-quarters of the clinical manifestations related to CGD began in the first year of life, which is in agreement with other works. Thus, for Mouy et al 20 and Wolach et al, 26 these clinical manifestations began before 1 year of age, respectively, for 70.8 and 65.7% of their patients. Köker et al 29 reported that, 82.4% of XLCGD and 50% of AR CGD developed such clinical manifestations before age 1 year.
However, appearance of the clinical characteristics of the CGD, even if they are early, does not constitute a guarantee for a positive diagnosis of this disease. Thus, in our study, we reported the case of a patient for whom the diagnosis of CGD was made at age 57 years. Some authors have reported cases of late CGD diagnosis in adulthood.36,48-52 Therefore there is a need to keep the possibility of a CGD diagnosis in mind even in an adult patient with recurrent purulent infection or granulomas.
Delay Between Onset of Clinical Manifestations and Positive Diagnosis
In our study, median age at diagnosis was 19 months (range:1-683). Positive diagnosis of CGD before 1 year was posed in 39% of cases, while the major percentage of positive diagnosis (44%) was posed between the age of 1 and 5 years. Only 1 case of extreme diagnosis was made a 57-year-old patient. Rawat et al 30 reported a median age at diagnosis of 36 months (range: 7-192), Martire et al 24 reported that the diagnosis of CGD was carried out in 20% of patients before the age of 1 year and that between 1 and 5 years, 10 and 20 years, respectively, 60% and 18 % of patients had been diagnosed, and an only 1 patient was diagnosed at 38 years of age. Winkelstein et al 2 reported that 76% of their patients were diagnosed before the age of 5 years, and 4% beyond the third decade of their life, the oldest patient diagnosed was 69 years old. Whereas van den Berg et al 28 reported that over half of their patients have had CGD diagnosis between 1 and 5 years of life, with 3 cases diagnosed after 40 years.
For our patients, time limit between clinical symptoms onset and CGD diagnosis was 10 months in median (range: 0-135). This time limit was of 10 months (range: 0-39), and also 10 months (range: 0-135) for patients born, respectively, after and before year 2000, no statistically significant difference was found between this 2 groups. A median delay in diagnosis of 2 years (range: 0-9), was reported by Rawat et al. 30 de Oliveira-Junior et al 32 reported that there was a substantial interval between the manifestation of the first symptoms of CGD and the time of diagnosis. The mean age at the first occurrence of symptoms was 23.9 months (range: 0.13-156), and the mean age at diagnosis was 52.7 months (range: 1-199). Whereas, Magnani et al, 53 reported an age at disease onset of 0.3 years (range: 0-9.2), and an age at diagnosis of 1.4 years (range: 0-12.8). Bortoletto et al 31 have observed a decrease in age at diagnosis in relation to more recent diagnostic dates, which would confirm better diagnostic tests and an accumulated awareness of CGD. These facts were confirmed by Zhou et al, 34 who reported a 5.5 months (range 1.2-16.5) delay, in their series, where 85.4% of patients were born after 2008, with an age of onset disease from 4 to 48 months.
The age at positive diagnosis was earlier in XL forms than in AR forms2,21,22,27,28,30,34,38 suggesting earlier and more severe expression of the XL forms. This fact probably reflects a milder phenotype of AR CGD, and hence more specific immunological testing at a early age than for X-linked forms, and not a bias in more early CGD diagnosis. Or it could be a more adequate screening of possibly affected boys in extended families with XLCGD, leading to very early diagnosis (eg, at birth) before any manifestations of disease signs. 28 As CGD is more commonly diagnosed in boys than in girls, we emphasize that girls with AR CGD may be missed as the index of suspicion is lower. This was certainly true for SCID, where x-linked disease was thought to be a higher percent of overall disease prior to newborn screening. In actuality girls were more likely to die undiagnosed because SCID was not considered, the same seems true for CGD.
In our study, 27% of patients had delayed weight development and hepatosplenomegaly at CGD diagnosis. What was reported by many authors22,26,27,30,31,34,36 and probably constituted a clinical consequence of recurrent infections and inflammations affecting the digestive tract. Hence the importance of thorough clinical examination and exploration of unexplained delayed height-weight development and unexplained hepatosplenomegaly.
Paraclinical Data
Anemia was found in 70% of our patients. This anemia was most often hypochromic microcytic probably related to a deficiency of supply or malabsorption, or secondary to inflammation. Wolach et al 26 reported that 74% of their patients were suffring from anemia. Kutluğ et al 54 found that anemia was one of the most common laboratory findings found in CGD. For Köker et al 29 the most common chronic manifestation in patients with CGD was anemia caused by chronic infection. According to Ross, 19 iron-deficiency anemia can be in some cases, a presenting symptom of CGD, and it constitutes one among its inflammatory and structural complications. 55
Hyperleukocytosis and thrombocytosis were found in 49% of our patients. Carnide et al 56 reported that hyperleukocytosis may be frequent even in the absence of acute infection, probably because of the persistence of a chronic stimulus. As for thrombocytosis, it is related to the inflammatory state of the patients. Thrombocytopenia would be related to the autoimmune manifestations of the CGD.57,58
Hypergammaglobulinemia account among the frequent inflammatory and structural complications of CGD and it’s considered among laboratory results frequently found in CGD patients. 55 IgG and IgA elevated values were found for, respectively, 53% and 49% of our patients, Rawat et al 30 reported such an elevation. Hypergammaglobulinemia would be related to the involvement of healthy effectors of acquired immunity in order to compensate for the failure of innate immunity. It’s also possible that a continuous inflammatory response causes chronic anemia and hypergammaglobulinemia in these patients.55,59 This hypergammaglobulinemia has been reported since the first studies on CGD, 60 so it should represent an alert for CGD diagnosis, especially in the presence of an evocative clinical picture. 56
Only 34% of patients for whom lymphocytes count was made showed an elevated TCD8+ count (c). Whereas Rawat et al 30 found that lymphocyte subsets were within normal range in most of their patients. As the Th1 type cells secrete IFN-γ which activates macrophages and TCD8 + subpopulation; these last can in turn amplificate to compensate for the macrophages failure.
No lymphocyte proliferation disorder was described during CGD, however, a defect in the regulation of T lymphocyte activity may be one of the causes of the hyperinflammatory state observed in the CGD. 19 Absent proliferation against specific antigens is likely to be the consequence of a lack or exceedance of the vaccination recall period. In all cases, a negative test should be confirmed after the vaccine recall. 61
In our study determination of CGD inheritance mode was made thanks to the study of patients’ pedigree which showed a predominance of the AR inheritance mode. Other populations, Iranian, 38 Palestinian and Israelian, 26 Tunisian, 62 Turkish 29 and migrant groups from North Africa and Arab countries in the European study, 13 showed the same results. Whereas, the XLform was more frequently found in French, 20 English, 27 Japanese, 10 American 31 and Latin American 32 studies.
Anti-Infectious Prophylaxis
The mainstay of clinical care in children with CGD is lifelong antibiotic and antifungal prophylaxis. Trimethoprim/ sulfamethoxazole (TMP-SX) (co-trimoxazole) is commonly employed for long-term prophylaxis. 16 Prophylactic TMP-SX reduced the frequency of major infections from 1 episode every year to 1 every 3.5 years in a study by Margolis et al. 63 Itraconazole is the recommended prophylaxis for fungal infections, since it’s highly effective against Aspergillus species. 16 Experiments have shown an improvement in phagocyte function following IFN-γ injections in-vitro, whereas, studies are still contradictory about the real benefit of using this therapy.64,65 Eighty-six percent of our patients have received prophylactic treatment with co-trimoxazole and itraconazole and only 22% received INF-γ not as prophylactic therapy but following severe infections.
Infectious Complications During CGD Monitoring
Patients with CGD suffer from a variety of recurrent bacterial and fungal infections, which occur most commonly in organs in contact with the outside world/: lungs, gastrointestinal tract, skin and lymph nodes that drain these structures. Because of both contiguous and haematogenous spread of infection, a wide range of other organs can be affected, most notably the liver, bones, kidneys and brain. In approximately two-thirds of patients, the first symptoms of CGD appear during the first year of life in the form of infections, dermatitis (sometimes seen at birth), gastrointestinal complications (obstruction or intermittent bloody diarrhea due to colitis).7,19
Microorganims Isolated From Patients
The infectious agents most frequently encountered during infectious complications are catalase positive, as Staphylococcus and Salmonella. Staphylococcus predominated in mucocutaneous and lymphatic infections. While Aspergillus, a fungus family member were mostly isolated in pneumonia.
In the revised definitions of diagnosis of invasive fungal disease (IFD), EORTC / MSG criteria 66 classifies these infections as: “proven,” “probable” and “possible.” These criteria include host, clinical, mycological and immunological conditions. The category of “possible” IFD was retained but was defined more strictly to include only those cases with the presence of appropriate host factors and with sufficient clinical evidence consistent with IFD. Whereas patients with CGD were included in such group even if their infection was without mycological support, which makes easier the diagnosis of possible “invasive fungal infection” in this patients.
In our study, Aspergillus antigenemia was only positive in 31% of cases, almost as reported by Beauté et al 67 The study of Aspergillus antigenemia by the galactomannan enzyme immunoassay is considered to be insensitive in CGD cases, because the antifungal prophylaxis and because the absence of fungal angio-invasion in this disease. 68 So, microbiological diagnosis of invasive aspergillosis remains difficult to establish in CGD.7,34 As percentage of infections or as percentage of infected patients, pulmonary, mucocutaneous and lymphatic infections predominate in the majority of studies (Table 1). Aspergillus, Staphylococcus and Salmonella representing the organisms most frequently found in these infections (Table 2). For our patients, Salmonella was the most frequently isolated during septicemia. Moreover, other unusual organisms make increasingly their apparition in these patients, hence the importance of carrying out a microbiological diagnosis each time before any anti-infectious treatment. 24
Granulomatous and Inflammatory Complications
Not all encounters with microorganisms lead to overt pyogenic infections in CGD. In some cases, chronic inflammatory cell reactions consisting of lymphocytes and histiocytes develop. These inflammatory reactions can then organize to form granulomas, one of the hallmarks of CGD which can cause various clinical symptoms of obstruction. Recent research has concentrated on the origin of the hyperinflammatory state of CGD patients, and 2 different causes which are not necessarily mutually exclusive have been proposed. These are the involvement of autophagy in the microbial killing process and a mechanism of excessive T-cell activation prevention by phagocytes. 19 On the other hand, efferocytosis (made by neutrophils) which is critical for quenching engoing inflammation and tissu necrosis, seems to be impaired in murine model of CGD.69,70
For our patients, such complications were relatively rare and limited, and were mostly present as clinical manifestations revealing CGD. During disease evolution, hepatomegaly and splenomegaly was found, respectively, in one-quarter and one-third of our patients. A genital granuloma was also found in 1 patient, and 1 patient has had a Crohn-like colitis. No patient had autoimmune complications and 6 patients have had atopic eczema lesions. Some authors 2,28,36,53 reported higher percentages of such complications, what is perhaps secondary to a more accurate clinical and paraclinical monitoring and/or a longer survival of patients.
Patients’ Evolution and Life Quality
Infections remain the major cause of death in CGD patients2,10,21,22,24-31,33,35,36,71,72 For our patients, the most frequent complications incriminated in deaths were pulmonary infections and septicemia. Aspergillus and Salmonella were the most implicated organisms. These results are consistent with those of other studies (Table 3). For Aspergillus, some species may be more virulent, as Aspergillus nidulans, who is more associated with invasive aspergillosis, because of its’ resistance against antifungal therapy. 73 Other organisms as Burkholderia cepacia formerly (Pseudomonas cepacia), are increasingly associated with morbidity and mortality in patients with CGD. This is due to the pathogenic mechanism of this bacterium, which induces significant necrosis levels of neutrophils, suggesting another severe pathological mechanism of the infection seen with Burkholderia in this patients. 74
Treatment with IFN-γ showed no improvement in our patients’ survival (Figure 6). Other studies have come to the same conclusions.22,24-26,28 Whereas most European physicians do not use IFN-γ prophylaxis routinely in CGD. 16 American studies show a greater benefit for the survival in the treated patients,2,31,64 although the American Academy of Allergy, Asthma & Immunology (AAAAI) guidelines recommended IFN-γ only for patients who have experienced severe infections.
A Karnofsky score greater than or equal to 80% was found in 59% of our surviving patients. Suggesting that CGD is a disease that can be associated with a satisfactory quality of life and that its adequate management makes it a chronic somewhat weakly disabling disease in some patients.
Most data from the literature agree that the association between GCD and malignant complications is unusual. None of our patients developed a tumor complication or hemopathy, despite the duration of the development of the CGD in some of them. However, rare cases of malignancies were reported. Wolach et al 26 found T-cell acute lymphoblastic leukemia (ALL) in a patient with XLCGD, which had a good evolution after treatment. The same authors 71 found a cervix carcinoma of the uterus in a 25-year old woman with AR-CGD. Other tumor types have also been reported as, malignant melanoma,6,28 cervical fibrosarcoma, 6 retinoblastoma, rhabdomyosarcoma 75 and Hodgkin lymphoma. 76
Conclusion
Our study is in accordance with previous studies that have shown pulmonary infections to be the most frequent clinical presentation followed by skin infections and lymphadenopathy, these complications were also the most frequent as CGD revealing manifestations. As reported, overall survival with respect to age in the AR forms of the disease was significantly higher than this in forms linked to X. Despite its disabling character, CGD remains one among PIDs most associated with a decent quality of life. Early diagnosis, based on thorough interrogation and complementary explorations, the introduction of anti-infectious prophylaxis and the initiation of adequate management, as soon as a complication is perceived, promotes pretty good evolution for this disease.
Author Contributions
FM: contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
HK: contributed to design; contributed to analysis and interpretation; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
ML: contributed to design; contributed to acquisition and analysis; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
MBK: contributed to design; contributed to acquisition; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
SR: contributed to design; contributed to acquisition; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
EBF: contributed to design; contributed to acquisition; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
MO: contributed to design; contributed to acquisition; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
MRB: contributed to conception; contributed to interpretation; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
MB: contributed to conception and design; contributed to analysis and interpretation; drafted manuscript; critically revised manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Footnotes
Acknowledgements
All data reported in this manuscript whether it be clinical, paraclinical and biological are part of the routine exams made for such patients. There is no conflict of interest of any other kind.
The authors would like to thank all the staff who have contributed to the monitoring and care of patients during these long years. We would also like to thank the patients and their families (especially parents), who were able over time to learn how to take care of their children.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.