
Abstract
To evaluate Tonsitin (10%
Introduction
Recurrent tonsillitis (RT) is a chronic inflammatory process of the palatine tonsils. Criteria for RT are: 4 to 5 or more episodes of true tonsillitis a year, symptoms for at least a year, with streptococci group A infection diagnosed in one of them.1,2
The common treatment approach of RT is systemic use of antibiotics. 3 In addition, supportive therapy could include oral or intramuscular steroids, analgesics using non-steroidal anti-inflammatory drugs as well as mouthwashes and lozenges. Recurrent tonsillitis could cause irregular tonsils’ size leading to upper respiratory disturbances. When there is no improvement in these RT conditions, tonsil removal (tonsillectomy) is the treatment of choice.2,4,5 It is among the most common surgical procedures performed in children in the United States 5 although its effectiveness is still controversial as well as the debate about the risks associated with the surgical procedure. 6
Palatine tonsils are a part of the mucosa-associated lymphatic tissue, a specialized compartment of the immune system that serves as a first line of defense against harmful environmental factors, including pathogenic microbes. 7 A comparative histology study of tonsils by Zhang et al 8 showed that reactive lymphoid hyperplasia and presence of chronic inflammatory cells, predominantly lymphocytes, are the two most important features seen in patients with RT.
In the current pilot study, we propose a new approach for treating RT using Tonsitin (10%
Lactic acid is a nontoxic constituent of the body fluids, which results as a normal intermediary of mammalian metabolism. It arises from glycogen breakdown, and from amino acids and dicarboxylic acids metabolism. The amount of endogenous lactic acid in human blood is about 90 mg/L (1-2 mmol/L) in resting condition. 9 Exogenous lactic acid usage ranges from its use as a food additive usually as a pH regulator, a preservative, and an antioxidant, to its use in different pharmacological preparations, such as parenteral/intravenous (IV) solutions, and acidification of the vaginal environment in women suffering from recurrent vaginal infection.10-13 Acidification of the vaginal environment with lactic acid supports the growth of “good” lactobacilli, which make up the essential part of the vaginal flora. At the same time, acidification slows the reproduction of harmful pathogens. Lactic acid-based preparations are thus used as a preventative in women who are susceptible to recurrent vaginal infections. 13 The European Food Safety Authority (EFSA) has published an opinion enabling the use of lactic acid as a decontaminating agent (antimicrobial processing agent) for beef. 14
Based on the above information, we proposed a new approach for treating RT using Tonsitin as a treatment for RT that might eliminate the need for repeated antibiotic treatment or surgery for patients with RT. The aim of this study was to determine the safety, feasibility, and efficacy of oral application of 10%
Methods
Ethics
The local hospital Helsinki ethics committee (EC) of the Shamir Medical Center, Zerifin, Israel, reviewed the study protocol that was granted a conditional approval, subject to the additional approval by the National Helsinki EC of the Israeli Ministry of Health (MOH). The MOH National Helsinki ethics committee issued its final approval letter for the study. The “Clalit” Health Services, Community Division Helsinki EC, reviewed and approved the study protocol.
Objectives
The primary endpoints of this study were to determine the safety and tolerability of Tonsitin (10%
The secondary endpoint of this study was to obtain preliminary efficacy assessment data of Tonsitin (10% lactic acid) treatment versus placebo by evaluating the following outcomes: changes in tonsils size grading, changes in frequency of tonsillitis episodes, and changes in quality of life (QoL).
Safety analysis was restricted to physical examination and AEs reporting only. No blood clinical laboratory assessment was done in this feasibility protocol.
Study Design and Plan
This was a prospective, randomized, double blind, placebo-controlled, 2:1 treatment groups clinical pilot study, to evaluate the safety, tolerability, and preliminary efficacy of 10%
The study was conducted at the Department of Otolaryngology, Head and Neck Surgery, in collaboration with the clinical pharmacology and toxicology unit at Assaf Harofeh Medical Center Zerifin in Israel, and was expanded to 2 additional outpatient Pediatric Community Ambulatory Clinics of “Clalit” Health Services in Petach Tikva, Israel. Children suffering from RT were also referred to the clinical sites above by other medical centers and physicians knowledgeable about the study.
All children enrolled were required to have a documented history of at least 4 tonsillitis episodes during the preceding year to study enrolment. Children suffering from RT identified as “predisposed” were approached, and their guardians were requested to sign an Informed Consent Form (ICF). No subject was enrolled in the study without obtaining the informed consent.
Overall, 51 children were recruited and allocated to 2 treatment groups (Tonsitin treatment and placebo) based on a prepared in advance randomization program, at a ratio of drug to placebo-treated patients of 2:1.
In general, the study consisted of a screening enrollment baseline visit, a treatment period, followed by a post-treatment follow-up period. By the end of the screening visit (visit 0-baseline), eligible children were provided with the investigational product (IP) Tonsitin syrup or placebo treatment to be taken PO (per os) twice daily for a month (morning and evening). During the first month of the treatment period, children were observed for tonsils size grading and pain assessment (visual analog scale [VAS]) biweekly at 14 and 30 days after the screening visit (visits 1 and 2, respectively). Following an intermission of 2 weeks, the treatment is renewed for another 2 weeks (a boosting treatment period), which at its end, the subject is required to come back again (visit 3) for tonsils size grading and pain intensity assessment (VAS). Following visit 3, the end of treatment (EoT) visit, children were observed for a post-treatment follow-up period of 6 months at 90 and 180 days post-EoT (see Figure 1 for study visit scheme).
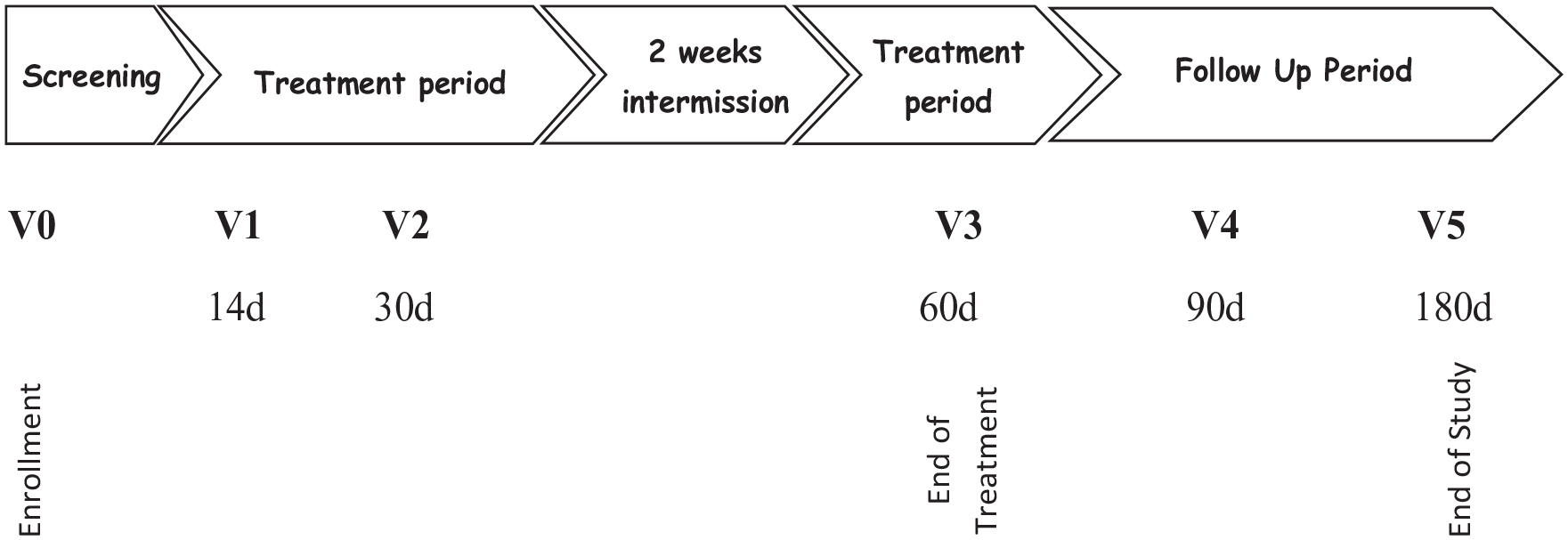
Study visit scheme.
Treatment safety, AEs, tonsils clinical presentation, and pain intensity were to be recorded throughout the whole study period duration. Overall, under the study protocol, each subject was involved in a total of 5 visits during study duration of about 8 months. Upon termination of the last follow-up visit (visit 5), the subject was considered to have completed the study. An additional telephone follow-up visit was added to collect data regarding reoccurrence of tonsillitis episodes during the 6-month period after end of study (EOS) in the study participating children.
Study Patients
The children suffering from RT who participated in the study were either children who were approached because they were identified on the patients’ lists of the involved clinics or children who were referred by other medical centers and physicians knowledgeable about the study. The pre-screening of participants was performed by referring physicians using the medical records of the clinic and only presumably eligible patients were screened. All screened children, a total of consecutive 51 children were found to be eligible based on compliance with inclusion/exclusion criteria at the screening visit.
The intention to treat (ITT) efficacy population included all 51 children who suffered from RT were enrolled into this study and were treated with oral administration of Tonsitin (10%
Inclusion/Exclusion Criteria
Included in our study were children at the ages of 5 to 16, suffering from recurrent documented sore throat with at least 4 tonsillitis episodes during the preceding year (at least, one verified by culture or rapid antigen testing for Streptococcus A), and with tonsils size graded between 2.5 and 4. Patient’s guardian has signed informed consent. We excluded children who were known as Streptococcus pyogenes carriers (based on positive throat culture or rapid test to S pyogenes in asymptomatic children), children requiring any other medication (topical or systemic) that may affect the course of the disease during the study period (eg, antibiotics, sedating antihistamines), children with known hypersensitivity to lactose, children suffering from an active peptic ulcer or from any concomitant disease, and children currently participating in another clinical study.
Statistical Methods and Sample Size
To establish the primary and secondary endpoints of this study protocol, safety and preliminary efficacy information were to be collected from 60 children suffering from RT who have enlarged tonsils, in a prospective, randomized, double-blind, placebo-controlled, 2 patient treatment groups, multicenter study design.
The 60 children suffering from RT were allocated into 2 study treatment groups (treatment vs placebo), at a ratio of 2:1, namely, 40 children were to be administered with the IP Tonsitin (10%
All measured variables and derived efficacy parameters were tabulated by descriptive statistics. This is to include summary tables providing: for categorical variables—sample size, absolute, and relative frequency by study group and for continuous variables at each time point, arithmetic mean, standard deviation, median, minimum, and maximum. For each efficacy, variable changes from previous visit and relative changes from baseline were performed.
The following statistical tests were used in the analysis of the efficacy data presented in this study: Two-sample t test for independent samples was applied for testing the statistical significance of the difference in relative changes in tonsils’ sizes at each visit between the study groups. The MMRM (mixed-effect model for repeated measures) model was applied for analyzing the difference between the study groups in tonsils’ sizes at treatment period and at follow-up period, adjusted by tonsil size at baseline.
Logistic regression was applied for analyzing the difference between the study groups in disease recurrence. Poisson regression was applied for analyzing the difference between the study groups in number of disease recurrences. Due to the small number of cases, Fisher’s exact test was used to analyze the difference between the study groups in the number of cases of disease appearance during the study treatment period.
The whole performance analysis was applied both on the ITT population and on the per protocol (PP) population. All tests were 2-tailed, and a P value of 5% or less was considered statistically significant.
The data were analyzed using the SAS® version 9.3 (SAS Institute, Cary, North Carolina).
The Study Product
The IP, 10%
All study IP had to be handled strictly in accordance with the study protocol and the container labels. Storage and dispensing instructions, and expiration date were supplied with the study medication upon delivery. The study medication was stored in a limited access area in a locked cabinet under appropriate environmental conditions (room temperature), until provided to the patient. Only authorized study personnel had access to the study IP.
Each individual patient treatment package kit consisted of ready for use 3 bottles to be provided to the individual patient of either treatment study group on the corresponding medication dispensing visits. The treatment dose was 5 ml twice daily of 10%
Safety, Tolerability, and Tolerability Assessments
Safety evaluated in terms of AEs, and tolerability in terms of compliance by the treated children. Preliminary efficacy assessed by changes in tonsils size grading within treatment period and during follow-up period, frequency of tonsillitis episodes during the follow-up period, and changes in QoL following treatment.
Adverse events
All AEs whether observed by the investigator or reported by the patient, were evaluated by the investigator and documented. All AEs were categorized as either a serious adverse event (SAE) or non-SAE and handled accordingly.
An SAE is defined as “any experience that is fatal or life-threatening, is permanently disabling, requires inpatient hospitalization, or is a congenital anomaly, cancer, or overdose.” In case of an AE, the clinical investigators initiated appropriate treatment according to their medical judgment and decided whether to withdraw the patient from the study.
Compliance assessment
Each delivery of the individual patients’ treatment packages was required to be identified by the investigators and recorded. Upon completion of each treatment period, the used treatment bottles were to be returned to the site and the residual Tonsitin syrup quantity was to be measured and recorded on the treatment compliance log at the site. The treatment accountability and compliance logs were checked during the periodic monitoring visits.
Upon receiving the product and on each visit, patients and their guardians were instructed on following the study protocol and its specific daily dosing.
Tonsil size assessment
The clinical presentation of their tonsil size was evaluated at screening and during each one of study visits throughout the study treatment period and follow-up period, by employing the Brodsky and Friedman 0 to 4 grading scale accordingly. 15
Pain assessment
The pain intensity was assessed at the enrollment baseline visit (visit 0) and during each one of study visits throughout the study treatment period and follow-up period, based on the patient’s own perception of their pain and its intensity using the hospital VAS for pain (from 0—for no pain to 10—for agonizing pain).
RT and microbiology tests
All children screened, were evaluated at the screening baseline visit (visit 0) for the most common pathologic bacteria known in RT (Haemophilus influenza, Staphylococcus aureus, S pyogenes, group A beta-hemolytic S pyogenes (GABHS), Streptococcus pneumonia, Escherichia coli, and Enterobacter).
A throat swab was collected from the patient’s posterior pharynx and tonsillar surfaces by the investigator and subjected for the rapid Strep antigen detection test (RSADT). A second throat swab was taken for a throat culture, the diagnostic gold standard. All microbiology tests were performed according to the routine hospital microbiology lab procedures.
QoL questionnaire
The impact of tonsillar hypertrophy at baseline (visit 0) and Tonsitin treatment on the QoL of the participating children was determined at 30 days post-treatment initiation and at the EoS follow-up visit (visit 5). The study employed the QoL questionnaire suggested by de Serres et al 16 for measuring QoL in children with sleep disorders.
Body weight, physical examinations, vital signs
Body weight and complete physical examinations, including vital signs, consisting of heart rate (HR), systolic and diastolic sitting blood pressure (BP) respiration rate (RR), and body temperature determination, were conducted at screening visit (visit 0) and at each one of study visits throughout the study treatment period and follow-up period.
Results
Study Population
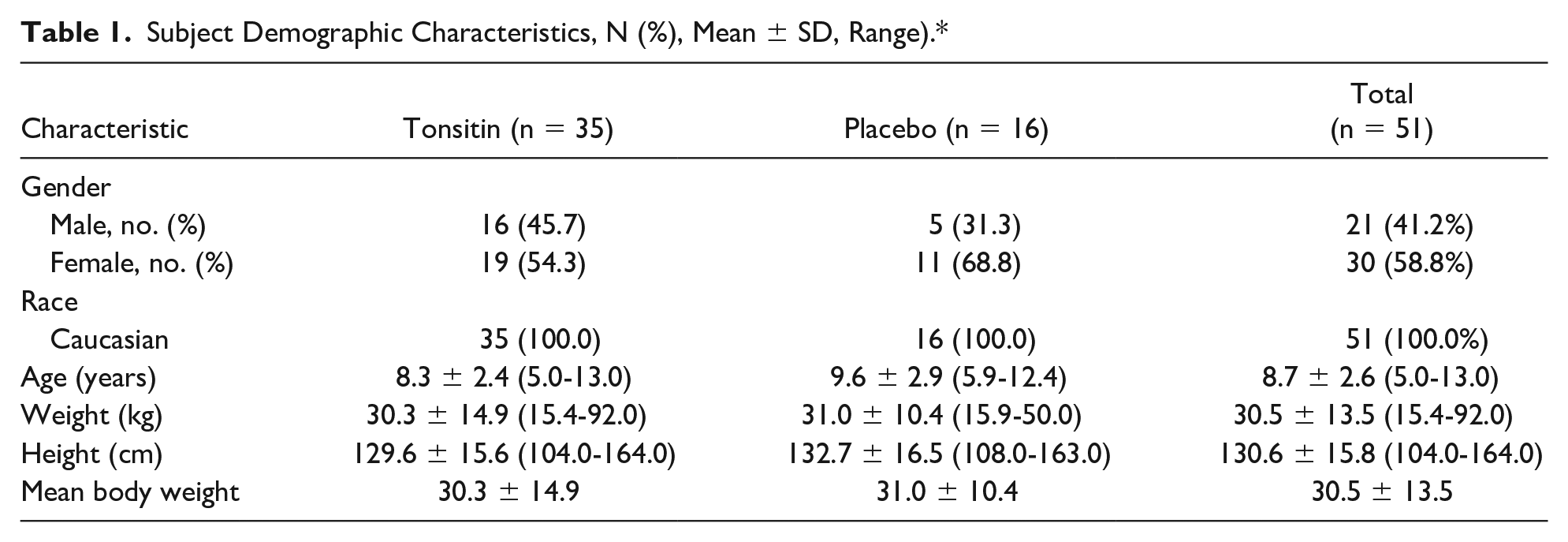
Between March 04, 2014 and November 19, 2015, 51 children who suffered from RT were enrolled into this study. The last final follow-up study visit was scheduled to July 13, 2016. Due to an unexpected low enrollment rate and the last retest date that covers the study batch IP stability, the study was terminated after enrollment of only 51 children. The demographics and characteristics of the ITT study population enrolled into the study are summarized in Table 1. Except for the proportion of females and males, which were not evenly spread among the study treatment groups, 58.8% and 41.2%, respectively, the treatment and the placebo groups were similar with respect to all the other demographic characteristics.
Subject Demographic Characteristics, N (%), Mean ± SD, Range).*
All the 51 children who were enrolled into this study were healthy children diagnosed as suffering from RT with at least 4 episodes in the preceding year to their enrollment (Table 2). All the enrolled children were healthy in regard to the other body system functions, and among the medical history and current medical condition.
Subject’s Clinical Characteristics at Screening—ITT Population, N (%), Mean ± SD, Range.
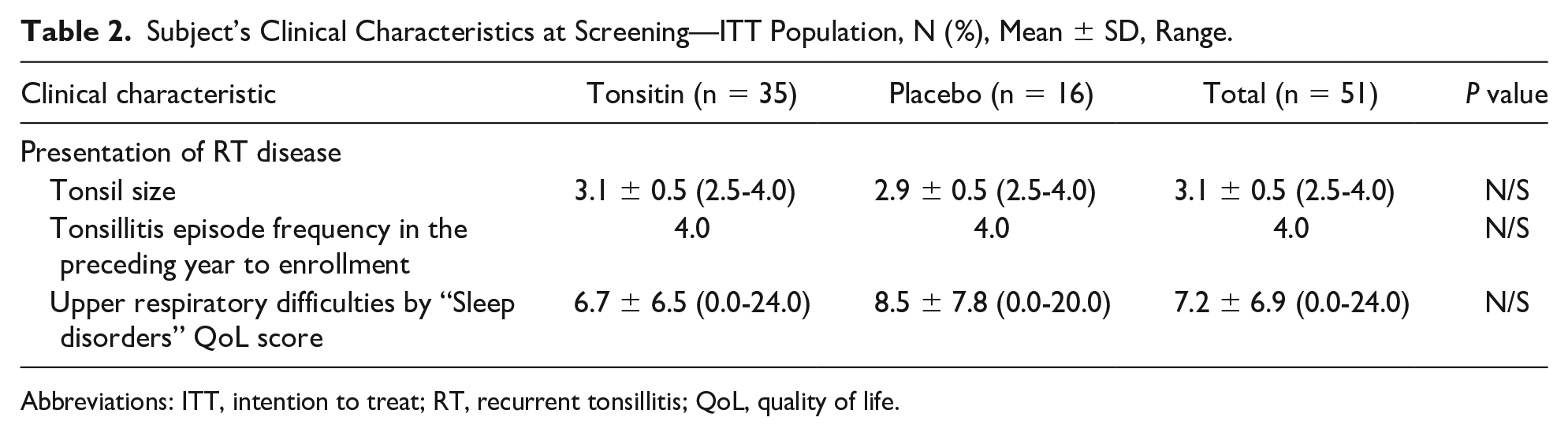
Abbreviations: ITT, intention to treat; RT, recurrent tonsillitis; QoL, quality of life.
Fifteen children identified had a premature termination. They were included in the safety analysis as well as in the ITT cohort since they received at least one treatment dose of the study IP.
Among the children identified as premature termination, 6 children, 3 of each study treatment arm had tonsillitis and/or pharyngitis treated with antibiotics (protocol dropout criteria). Five children, 3 of the study Tonsitin treatment arm and 2 of the placebo arm, were defined as treatment non-compliance. Two other children, one of each study treatment arm were protocol non-compliance. One child of the study Tonsitin treatment arm did not complete the study due to an SAE, and one child of the placebo arm had withdrawn consent.
The PP population consisted of the subset of 36 children from the ITT population who complied with the treatment regimen and completed all study visits as planned (ie, excluding dropouts).
It should be emphasized that since all enrolled children (n = 51/100%) received at least one treatment dose of the study IP, all children were considered ITT population (for safety and efficacy analysis).
Table 3 summarizes the study subject disposition by study treatment group. A total of 51 children (100%) comprised the safety and ITT study populations.
Frequency (N, %) of Subject Disposition.
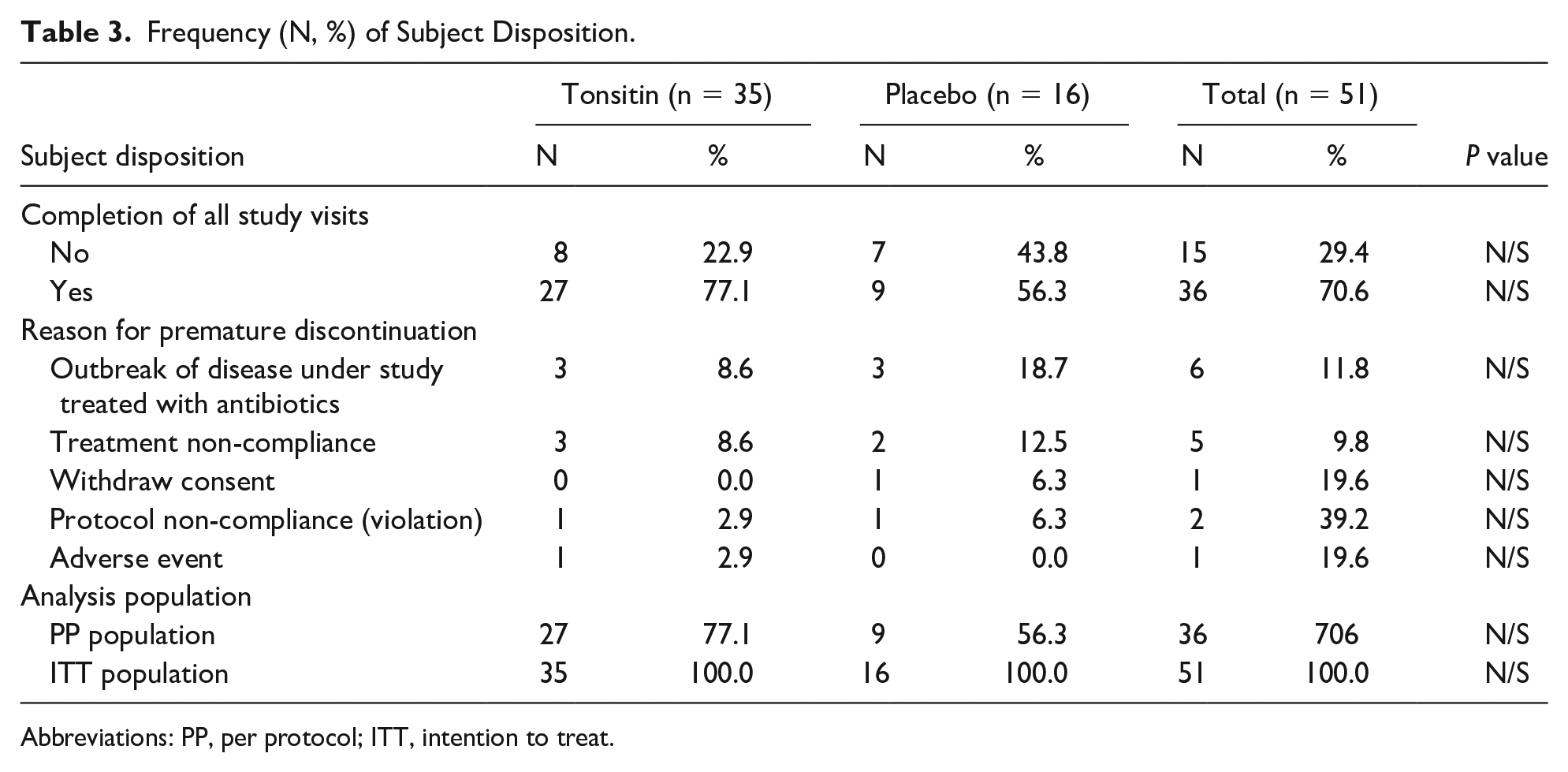
Abbreviations: PP, per protocol; ITT, intention to treat.
Treatment Compliance
Compliance with the study protocol daily treatment regimen was recorded and traced on the study IP accountability logs and checked by measurement of the returned volume in each treatment bottle at the EoT period.
Five children, 3 of the study Tonsitin treatment arm and 2 of the placebo arm, were defined as treatment non-compliance since they received only less than 20% (10-70 ml), of the total treatment dose and were excluded from the PP analysis.
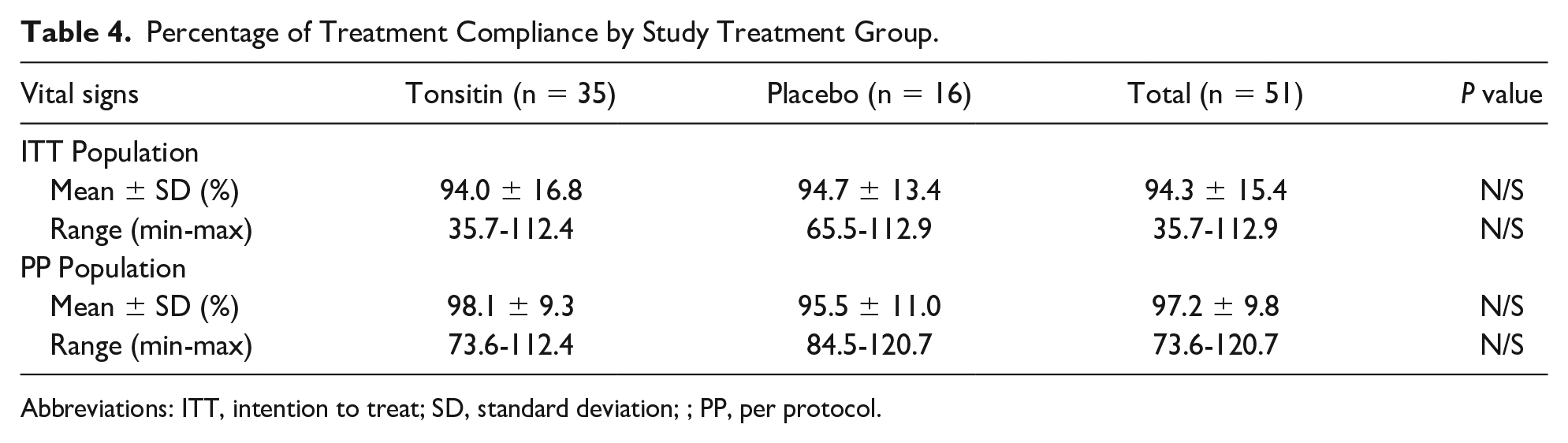
In general, the treatment regimen was tolerable among the participation children and a mean percentage treatment compliance of 94.0 ± 16.8% was recorded for the Tonsitin investigational treatment group of the ITT population with a slight nonsignificant higher percentage treatment compliance in the placebo treatment group (Table 4).
Percentage of Treatment Compliance by Study Treatment Group.
Abbreviations: ITT, intention to treat; SD, standard deviation; ; PP, per protocol.
However, much higher mean percentage treatment compliance data were assessed for the PP population reaching a mean percentage treatment compliance of 98.1 ± 9.3% and 95.5 ± 11.0% for the Tonsitin investigational treatment and the placebo treatment study groups, respectively (Table 4).
Safety Evaluation
The routine safety data that were followed throughout the study period, included: deaths, SAEs, physical examination, body weight, vital signs assessment (to include HR, BP, RR, and body temperature) performed at each visit during the treatment period and at 3 and 6 months post-treatment during the follow-up period visits.
Adverse events
No deaths occurred during the study period. Review of the mean vital signs and mean body temperature values, comparable with baseline, show that in general, except for small changes in vital signs and body temperature values that were observed at isolated study time points for specific individual patients, all vital signs and body temperature values were within the normal range without any clinical consequence and with no specific effect observed in one of the study treatment groups.
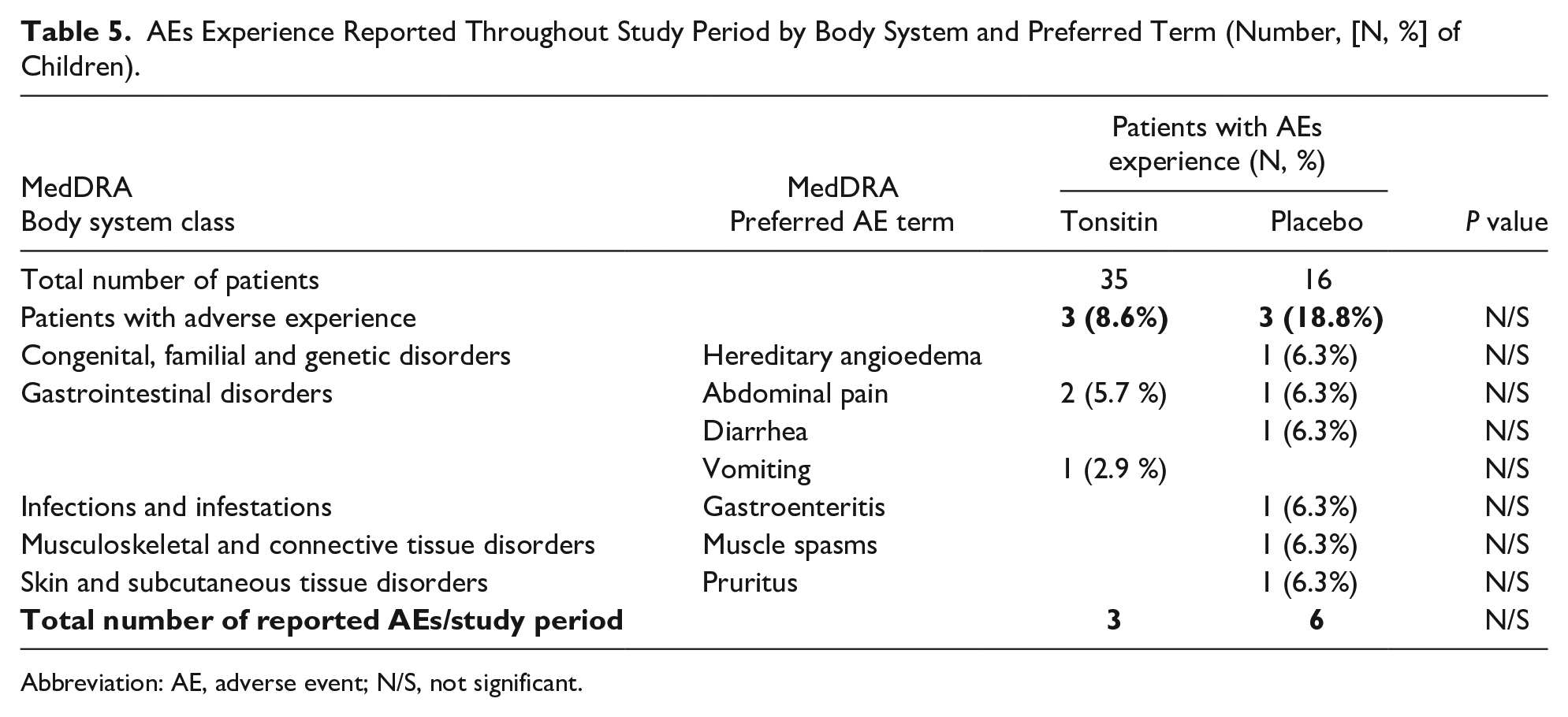
Overall, only 6 children (6/51 11.7%), 3 of each study treatment arm, had treatment-emergent reported adverse experiences (Table 5). A total of 9 cases of adverse experience were recorded in the 6 children enrolled. Among the 9 cases that were observed throughout treatment period or follow-up period, the most common events were reported for children with gastrointestinal disorders (4/51 7.84 %), 2 of each study treatment arm.
AEs Experience Reported Throughout Study Period by Body System and Preferred Term (Number, [N, %] of Children).
Abbreviation: AE, adverse event; N/S, not significant.
In regards to AEs severity, the majority (7/9 77.8%) of the 9 AEs that were reported throughout the Tonsitin or placebo treatment period were described as mild, one (1/9 11.1%) AE was described as moderate and only one AE (1/9 11.1%) was graded as severe, the hereditary angioedema of one of the placebo group patients.
Vital signs
Review of the mean vital signs values and the mean body temperature values, comparable with baseline (pre-Tonsitin treatment) show that in all vital signs values and mean body temperature values, were within the normal range without any clinical consequence and with no specific effect observed in one of the study treatment groups.
Body weight
Review of the mean body weight values comparable with baseline (pre-Tonsitin treatment) show that there was a slight non-significant decrease in the mean body weight values of the Tonsitin-treated children comparable with the placebo group during the treatment period. All body weight values were within the normal range without any clinical consequence and recovered during the follow-up period.
Physical examination
There were no clinically significant physical examination findings during the study.
Tonsil size
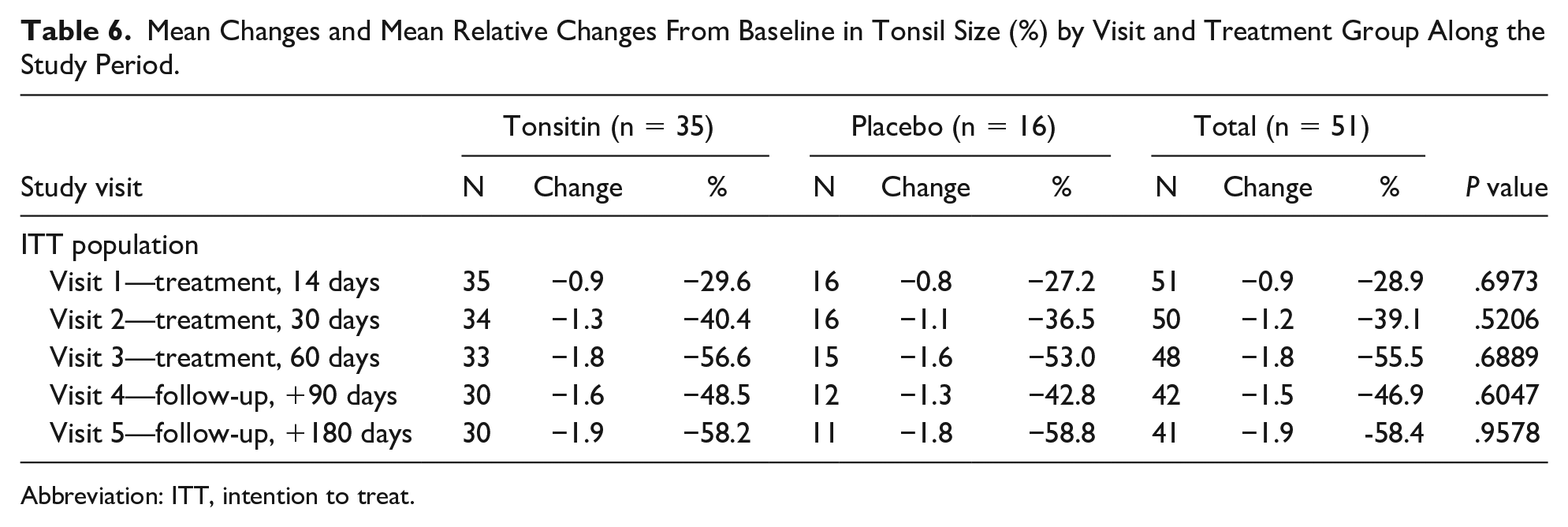
Tonsil size declined from 3.1 ± 0.5 (median: 3, min: 2.5, max: 4) and 2.9 ± 0.5 (median: 3, min: 2.5, max: 4) to 1.3 ± 0.7 (median: 1, min: 0, max: 4) and 1.3 ± 1.0 (median: 1, min: 0, max: 2.5) (P = .6889) at the EoT visit in the Tonsitin and placebo treatment arms, respectively. The decline in the mean relative tonsil size score continued for a much longer time after the treatment was completed and achieved its greatest change from baseline (–1.9 points in tonsil size score) during the study follow-up period observed at 180 days post-treatment, which accounts for 58.2 ± 29.6% and 58.8 ± 37.4% (P = .9578) mean relative decrease in tonsil size from baseline in the Tonsitin and placebo treatment arms, respectively (Table 6).
Mean Changes and Mean Relative Changes From Baseline in Tonsil Size (%) by Visit and Treatment Group Along the Study Period.
Abbreviation: ITT, intention to treat.
Tonsillitis episodes outbreak during the study treatment period
Among the children identified as premature termination, leading to their removal from the study were 6 children due to the onset of tonsillitis and/or pharyngitis that was treated with antibiotics. Three of these children were in the Tonsitin treatment group, while the other 3 were enrolled to the placebo control group (P = .3627).
Changes in frequency of tonsillitis episodes after study EoT
By the EoS, children who fulfilled the requirement of participation in the study for over a year were approached by an additional telephone follow-up questionnaire aimed to record the tonsillitis episodes’ appearance and their frequency following the study treatment. The information for 2 participating children, one of each study treatment arm was not available.
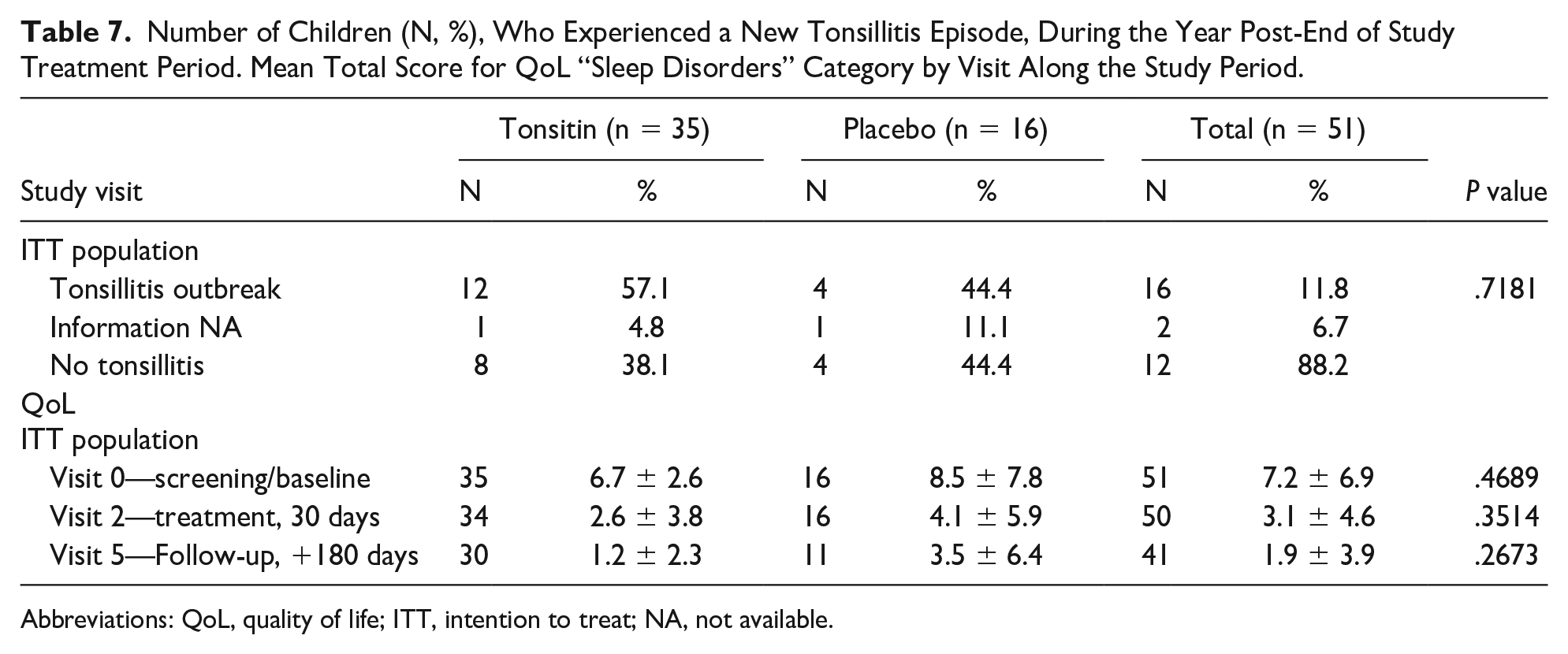
Table 7 summarizes the number of children who experienced a new tonsillitis episode during the year post-EoS treatment. Although the percentages of appearance of tonsillitis are in favor of the placebo group, the given P value indicates that these results are random and do not show an advantage of the placebo group (P = .7181).
Number of Children (N, %), Who Experienced a New Tonsillitis Episode, During the Year Post-End of Study Treatment Period. Mean Total Score for QoL “Sleep Disorders” Category by Visit Along the Study Period.
Abbreviations: QoL, quality of life; ITT, intention to treat; NA, not available.
Changes from baseline in QoL assessment
The impact of tonsillar hypertrophy at baseline (visit 0) and Tonsitin treatment on the QoL of the participating children was determined at 30 days post-treatment initiation and at the EoS follow-up visit (visit 5). The study employed the QoL questionnaire suggested by de Serres et al 16 for measuring QoL in children with sleep disorders.
Quality of life score was 6.7 ± 2.6 and 8.5 ± 7.8 (P = .4689) at the screening visit, 2.6 ± 3.8 and 4.1 ± 5.9 (P = .3514) during the treatment, and 1.2 ± 2.3 and 3.5 ± 6.4 (P = .2673) at the follow-up visit, for the Tonsitin and control groups, respectively. Total scores for each one of the QoL questionnaire categories along the study treatment and follow-up periods demonstrate a clear reduction from the initial baseline total score in all QoL questionnaire categories, indicating a growing improvement following treatment that continue throughout the follow-up period of 6 months post-EoT (see Table 7). The given P value represents that there is no significant difference between the mean total score at the study visits between the 2 study groups.
Discussion
Recurrent tonsillitis may be complicated by considerable morbidity and poor QoL. 17 There are several methods for treatment of RT. These can range from the use of broad-spectrum antibiotics to the need of surgical intervention.1,18
Recent studies found that local reaction and biofilm of bacteria microcolonies might lead to a reaction within the adenotonsillar that causes hyperplasia and sleep disorders associated with it.19,20 Such studies are in line with our rationale to treat adenotonsillar diseases conservatively. Our current pilot study rationale was using a conservative treatment for prevention of RT by oral application of 10%
The primary endpoint of safety and tolerability of this study was achieved alongside slight possible lactic acidosis symptoms and non-significant decline in body weight that requires additional investigation to rule out relation to the study medication.
In regard to the secondary efficacy study endpoints of Tonsitin treatment, there was a statistically non-significant reduction in mean tonsil size, which was noticeable during the treatment period; reaching a decrease in tonsil size of more than 10% compared with tonsil size in the first visit and becomes more moderate during the follow-up period (3 and 6 months post-treatment). This reduction in mean tonsil size continued far after the treatment was completed and could be observed at 90 and 180 days post-EoS treatment. On the other hand, the recurrence of tonsillitis and the frequency of tonsillitis episodes were recorded at a higher rate in our study, in the Tonsitin-treated children arm than in the placebo-treated, reporting a clear decline in tonsillitis recurrence rate up to their total disappearance years post-treatment. Yet, these study results were not statistically significant. Furthermore, there was a direct association between the reduction in tonsil size and the level of improvement in upper respiratory difficulties (airway obstruction) as reflected in the mean “sleep disorders” QoL questionnaire scores along the study treatment and follow-up periods. In this case also, there is full parallelism of the improvement in upper respiratory difficulties to the continuing tonsil size reduction effect for after the treatment was completed and it was documented at 90 and 180 days post-EoS treatment.
However, in this clinical study, our secondary efficacy results show that the remarkable improvement from baseline, in the tonsil size, and improvement in upper respiratory difficulties was demonstrated in both the Tonsitin and the placebo-treated study groups. Placebo was found to be as good as Tonsitin treatment for all of the studied clinical efficacy assessment endpoints, during the treatment period as well as at the EoS, 6 months post-EoT. Given the improvements in both study groups, one might suggest that perhaps these signs and symptoms simply get better with time. On the other hand, Tonsitin is safe to be studied in the future for other pediatric diseases.
It is worth to mention that the study submission was delayed of about 6 years after the last patient’s visit, due to the need of the sponsor to finalize the patent registration on the IP. Only when this process was completed we were able to send the manuscript for publication.
Study Limitations
The present clinical study results may be affected by: First, our decision to use an off the shelf product, food grade raspberry flavor concentrated syrup, as the “placebo,” limited the ability to accurately control the placebo product ingredients that may have contained citric acid, potassium sorbate, and sodium benzoate as preservatives affecting the placebo treatment arm results. Second, tonsillar hyperplasia is problematic since there is no objective procedure for quantifying hyperplasia of the tonsils or adenoids. A large tonsil that seems to one examiner to be physiological and of no clinical consequence may be judged by another to be pathologically enlarged need of treatment. Third, we cannot exclude the fact that different and much better results would have been achieved for the statistical analysis for recurrence of tonsillitis if the total number of tonsillitis episodes in the year prior to enrollment was documented and not only the last 4 tonsillitis episodes required as a minimum to be eligible by the study inclusion criteria. Fourth, the small sample size of the total patients. The study’s sample size was based on a .05 level for significance and not a .005 as was suggested by recent studies. 21 Our study was designed with the guidance of statistician during 2012 according to the statistical and clinical norms that were accepted at that time. The current attitude to lower the P value to .005 was not wide spread at that time.
Conclusion
In conclusion, in spite of the fact that the safety and tolerability of this study was achieved, the efficacy results achieved following Tonsitin (10%
Author Contributions
Dr Ahmed Taha, Dr Elkana Kohn, and Dr Yuval Mizrakli had primary responsibility for protocol development, patient screening, enrollment, outcome assessment, preliminary data analysis, and writing the manuscript. Dr Haim Gavriel, Dr Matatiahu Berkovitch, and Dr Yehezkeli participated in the development of the protocol and analytical framework for the study and contributed to the writing of the manuscript. Dr Shlamkvich contributed to patient screening. Dr Taha and Dr Berkovitch supervised the design and execution of the study. All authors approve this paper for publication.
Footnotes
Acknowledgements
The authors are grateful to the authors of the papers included in this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sponsors and Collaborators: Yali Pharmaceuticals.
Ethical Approval and Consent to Participate
The study was approved by the local and national Helsinki ethics committee.
Compliance With Ethical Standards
The study was conducted according to the guidelines of the Declaration of Helsinki. The informed consents were approved by the Helsinki committee.