
Abstract

Educational Objectives
In patients with classical 21-hydroxylase deficiency, the metabolic block in aldosterone and cortisol synthesis results in raised hormone precursor, 17-hydroxyprogesterone.
Infants with simple virilizing congenital adrenal hyperplasia do not produce cortisol efficiently; however, they can synthesize adequate aldosterone to maintain salt-water balance.
Case Report
A term infant was born via cesarean section to a gravida 1 para 0 woman from a non-consanguineous marriage. Antenatal history was uneventful. There were no known inherited disorders in the family. The infant was small for gestational age (SGA), having a head circumference of 32 cm, body length of 45 cm, and birth weight of 2.05 kg, corresponding to the 10th centile on the pediatric growth chart. At birth, the infant’s blood pressure was 70/40 mm Hg, pulse rate was 130 beats per minute, temperature was 37°C, respiratory rate was 46 breaths per minute, and oxygen saturation was 100% under room air. Physical examination revealed non-dysmorphic features, normotensive anterior fontanelle, good pulse volume, warm peripheries, and a capillary filling time <2 seconds. The cardiorespiratory system and abdomen were unremarkable. However, penoscrotal hypospadias and bifid scrotum were found on genital examination. During the first few hours of life, the infant appeared irritable and restless. Capillary blood glucose testing revealed a blood glucose concentration of 1.4 mmol/L (reference interval [RI]: 2.8-4.5 mmol/L) which improved upon intravenous dextrose infusion. Neonatal hypothyroid screening with cord blood thyroid stimulating hormone was 6.3 mIU/L (RI: 1.05-36.10 mIU/L).
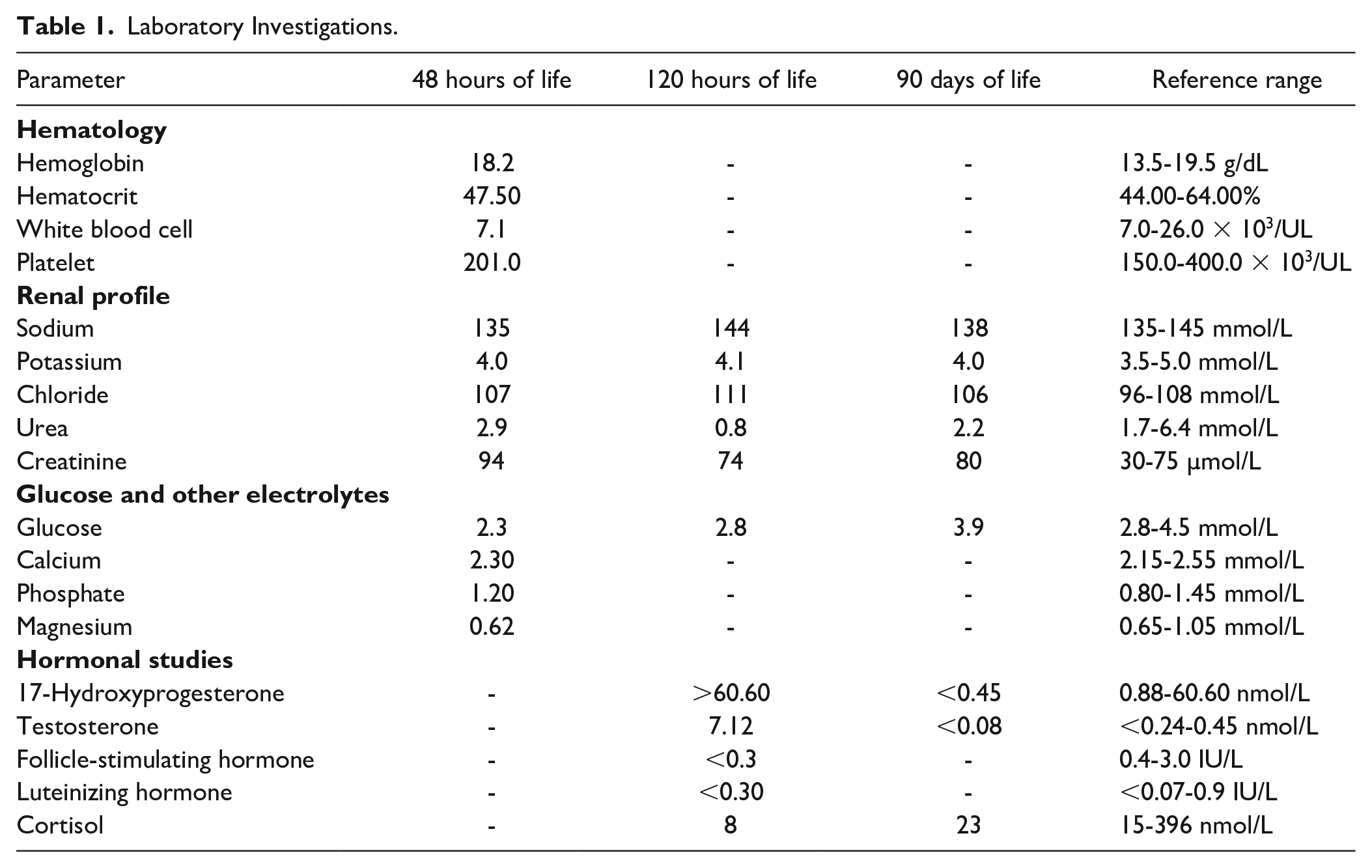
Hypoglycemia, which was persistent at 48 hours of life (Table 1), and the presence of abnormal genitalia prompted clinicians to perform further investigations. Full blood count and renal profile were unremarkable (Table 1). Serum follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were low, however, within pre-pubertal limits (Table 1). Morning serum cortisol was suppressed while serum testosterone and 17-hydroxyprogesterone (17-OHP) were elevated (Table 1). Chromosomal analysis revealed a 46XY karyotype with no chromosomal abnormalities, consistent with pelvic ultrasound findings of bilateral testes within the scrotal sac and absent uterus.
Laboratory Investigations.
Final Diagnosis
A diagnosis of simple virilizing (SV) congenital adrenal hyperplasia (CAH) was made based on the clinical and biochemical investigations. The patient was started on syrup hydrocortisone 0.5 mg 8 hourly (10 mg/m²/day) and syrup fludrocortisone 25 µg once daily (150 µg/m²/day). Blood workup during a clinic visit 3 months later revealed adequate suppression of 17-OHP and testosterone with normal blood glucose and electrolyte levels (Table 1). Currently, he continues his follow-up under the pediatric endocrine team and is planned for surgical correction of penoscrotal hypospadias and bifid scrotum.
Discussion
CAH encompasses several inherited adrenal disorders characterized by a mutation in one of the enzymes necessary for adrenal steroid biosynthesis. 1 The worldwide incidence of CAH is approximately 1:15 000 2 while the incidence in Asian populations is estimated to be around 1:44 000. 3 21-Hydroxylase enzyme deficiency (21-OHD) due to a mutation at the CYP21A gene is the commonest variant of CAH, accounting for more than 90% of all cases.1,4 In this disorder, a reduced conversion of progesterone to 11-deoxycorticosterone and 17-OHP to 11-deoxycortisol leads to aldosterone and cortisol deficiencies (Figure 1).
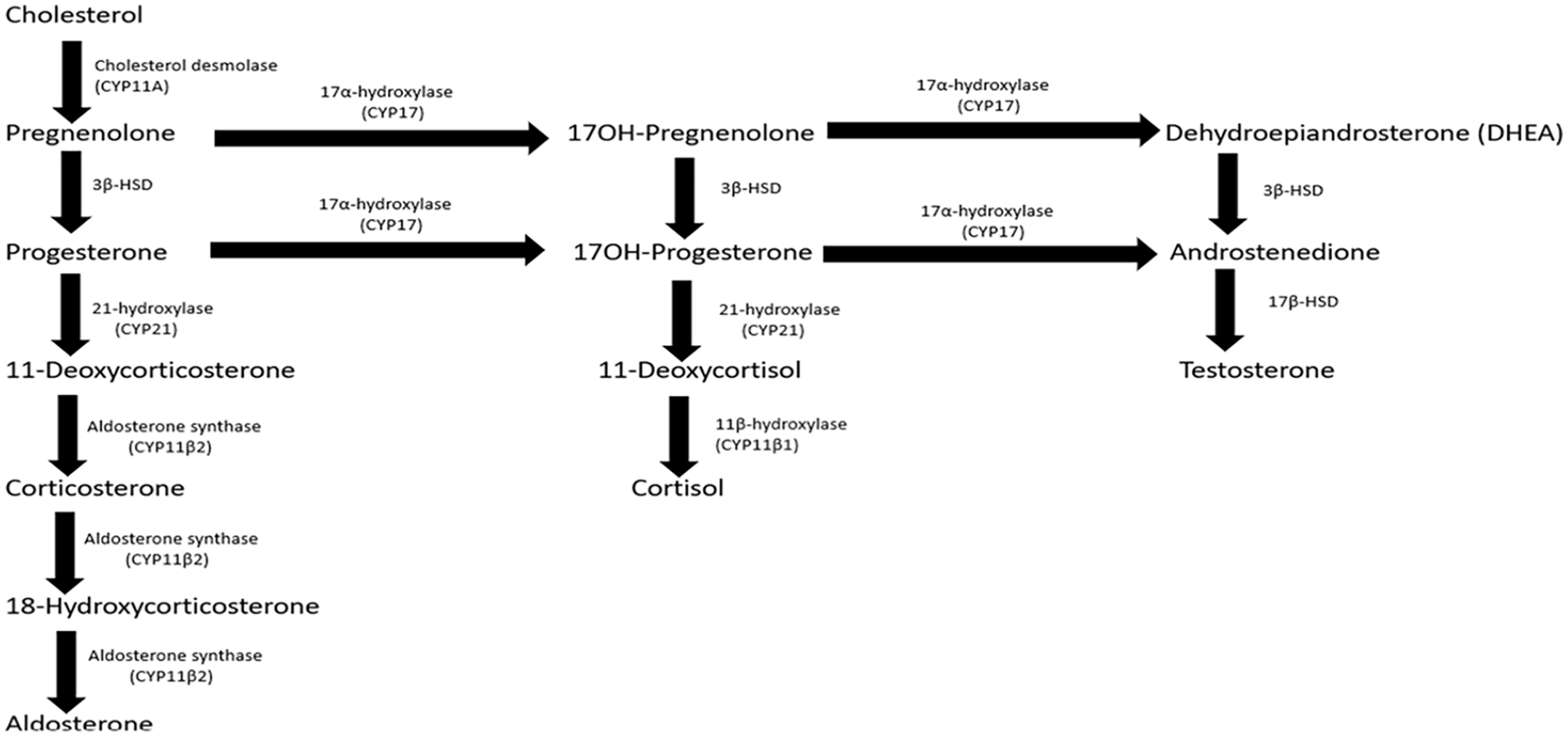
Pathways of adrenal steroidogenesis.
The spectrum of this disorder ranges from classic CAH, which is associated with complete enzyme deficiency, to non-classic CAH (NCCAH), which is associated with partial enzyme deficiency. 4 Patients with classic CAH can be further classified into salt-wasting (SW) and non-SW forms (also known as SV form) depending on the quantitative difference of 21-hydroxylase enzyme (21-OH) and clinical presentation. 4 Unlike the SW forms where complete mineralocorticoid deficiency is observed, our patient was clinically well hydrated with no signs of vascular insufficiency or hypotension and did not have typical biochemical findings of hyponatremia, hypochloremia, and hyperkalemia. This could be explained by the quantitative difference of 21-OH in the SV form, whereby as little as 1% of 21-OH activity permits aldosterone synthesis to prevent life-threatening SW. 5
Glucocorticoid deficiency was present in this patient, evidenced by low morning cortisol. Hypocortisolism decreases gluconeogenesis and reduces the antagonistic effect on insulin, preventing normal blood glucose regulation. 6 This explains the persistent hypoglycemia in our case, which continued even after the period of transitional glucose regulation at 48 hours of life. Small for gestational age infants, similar to ours, have lower glycogen and fat stores. This is another risk factor for hypoglycemia in this patient due to the limited ability to generate glucose via gluconeogenesis. 7
In healthy infants, 17-OHP levels rapidly decrease during the first few days of life. 8 However, in infants with CAH, the metabolic block in aldosterone and cortisol synthesis results in gradual accumulation of hormone precursors which are shunted into the androgen pathway 1 (Figure 1). Thus, the concentration of 17-OHP progressively escalates with time. 8 This may lead to a false negative result if sampling is obtained within the first 2 days of life, a time when 17-OHP concentration is still close to baseline. 4 It is for this reason that 17-OHP testing was performed on day 5 of life in our patient. Besides that, 17-OHP levels may also be raised in ill, stressed, or SGA infants who do not have CAH, 8 producing a false positive result. With reference to the 2018 Endocrine Society clinical practice guidelines, 17-OHP <6 nmol/L excludes 21-OHD, while a result >30 nmol/L as seen in our patient, confirms the diagnosis. 8
In CAH, an elevation in adrenocorticotrophic hormone (ACTH) from reduced cortisol feedback generates more precursors proximal to the enzyme blockage. 1 Being substrates for an unobstructed androgen pathway, these precursors are channeled to generate excessive androgen, 1 evidenced by increased testosterone in our patient. These elevated sex steroids may also induce negative feedback on the pituitary gland to decrease FSH and LH secretion. 9 Varying degrees of virilization and ambiguous genitalia are usually observed in the female CAH infant due to androgen excess, but not in the male. 1 Hypospadias, defined as a midline fusion defect of the penile urethra, albeit being one of the most common congenital anomalies in the male infant, is not commonly associated with 21-OHD. 10 Yet, a rare form of CAH, 3-beta-hydroxysteroid dehydrogenase deficiency (3β-HSD), can result in raised Δ5-steroid to Δ4-steroid ratio (pregnenolone to progesterone, 17α-hydroxypregnenolone to 17-OHP, and DHEA to androstenedione), ultimately leading to deficient testosterone production in both the testes and adrenal glands. 1 Severe forms of 3β-HSD are associated with manifestations of under virilization such as hypospadias, micropenis, undescended testes, and bifid scrotum. 11 Although molecular testing was not performed, it is less likely that our patient has this form of CAH because his testosterone level remained unsuppressed. 1 Under virilization of male infants with CAH is rare. 1 Similar to 2 other reported cases in the literature, hypospadias in our case may be attributed by abnormal Leydig cell function in utero triggered by unknown environmental chemicals, 10 or mutation of specific transcription factors such as mastermind-like domain-containing 1 (MAMLD1), HomeoboxA13 (HOXA13), or SRY type HMG box9 (SOX9). 12 These mutations can only be confirmed with advanced genetic analysis which is currently unavailable at our center.
The diagnosis of CAH in this patient was formulated on clinical and biochemical findings. He most likely has 21-OHD, as it is the commonest variant worldwide. 11β-hydroxylase enzyme deficiency (11β-OHD), accounting for approximately 5% of CAH cases, 1 is also a possible variant since it is not typically associated with SW. 1 Although 11β-OHD impairs the conversion of 11-deoxycorticosterone to cortisone and 11-deoxycortisol to cortisol (Figure 1), mineralocorticoid activity is not affected.1,13 This is because the primary enzyme mediating the conversion of 11-deoxycorticosterone to the final product, aldosterone, is aldosterone synthase1,13(Figure 1). An elevated 11-deoxycorticosterone and 11-deoxycortisol, though not routinely assayed in our setting, would be helpful to differentiate 11β-OHD from 21-OHD as their levels will be suppressed in 21-OHD. 1 17 alpha-hydroxylase deficiency (17-OHD), a rare variant of CAH associated with impaired conversion of pregnenolone to 17-hydroxypregnenolone, and progesterone to 17-OHP (Figure 1), is also unlikely in our case as patients generally present with suppressed 17-OHP and androgen levels. Due to resource and financial constraints, genotyping could not be conducted in our setting. Molecular testing involving techniques like Sanger sequencing or next-generation sequencing is currently used by many reference laboratories and should be considered when an equivocal adrenocortical profile is obtained even after cosyntropin stimulation or when genetic counseling is required. 8
Management of classic 21-OHD CAH in early infancy involves mineralocorticoid and glucocorticoid replacements, as demonstrated in our patient. Long-term glucocorticoid treatment inhibits the secretion of corticotrophin-releasing hormone and ACTH to reduce adrenal androgen production. 4 While patients with SV form of CAH are theoretically able to secrete low concentrations of mineralocorticoid to prevent SW, treatment with fludrocortisone, as seen in our patient, supports adrenocortical suppression and lessens the need for high-dose corticosteroids, which may cause Cushing syndrome. 4 The ultimate aim of CAH treatment is to prevent adrenal crisis, control androgen excess, and ensure normal growth and development. 8 Regular monitoring of electrolytes, hormone levels, and growth parameters is essential to optimize therapy and prevent complications. 8 In this case, normalized 17-OHP and stable glucose levels after treatment are crucial indicators of the effectiveness of this patient’s treatment.
Conclusion
This case report highlights the clinical presentation, diagnostic evaluation, and management of classical SV CAH in a newborn male. Although hypo-virilization features are rarely associated with male infants, this diagnosis was formulated based on other clinical findings and available biochemical investigations. The etiology of atypical genitalia in this child remains unknown, however possible hypotheses such as abnormal Leydig cell function in utero or mutation of specific transcription factors have been suggested. Early diagnosis and prompt initiation of treatment will help prevent complications, promote normal growth, and improve the long-term outcomes for this child.
Author Contributions
Footnotes
Acknowledgements
The authors(s) would like to thank the Director General of Health Malaysia for his permission to publish this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
This case report publication is exempted from ethical review and approval because the study or data collection is based entirely on data abstraction from existing medical or laboratory record, with no interaction with the human subject concerned and with no collection of identifiable private information. Verbal informed consent is obtained from the patient’s legal next of kin.