
Abstract
Each month, subscribers to The Formulary Monograph Service receive 5 to 6 well-documented monographs on drugs that are newly released or are in late phase 3 trials. The monographs are targeted to Pharmacy & Therapeutics Committees. Subscribers also receive monthly 1-page summary monographs on agents that are useful for agendas and pharmacy/nursing in-services. A comprehensive target drug utilization evaluation/medication use evaluation (DUE/MUE) is also provided each month. With a subscription, the monographs are available online to subscribers. Monographs can be customized to meet the needs of a facility. Through the cooperation of The Formulary, Hospital Pharmacy publishes selected reviews in this column. For more information about The Formulary Monograph Service, contact Wolters Kluwer customer service at 866-397-3433.
Indications
Daprodustat is a hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitor approved by the Food and Drug Administration (FDA) for the treatment of anemia due to chronic kidney disease (CKD) in adults who have been receiving dialysis for at least 4 months. Daprodustat is not indicated for use as a substitute for red blood cell transfusion in patients requiring immediate correction of anemia, or for treatment of anemia due to CKD in patients not on dialysis. 1
Anemia associated with CKD has been previously treated with erythropoiesis-stimulating agents (ESAs), including darbepoetin alfa, epoetin alfa, epoetin beta, epoetin omega, epoetin delta, and methoxy polyethylene glycol-epoetin beta. 2 However, all these drugs are given by injection. Daprodustat and the other HIF-PH inhibitors pending approval (eg, roxadustat, vadadustat) are given orally and may be an alternative to ESAs.3,4
AstraZeneca received a Complete Response Letter (CRL) from the FDA in August 2021 regarding its New Drug Application (NDA) for use of roxadustat in the treatment of adults with anemia due to CKD, in both non–dialysis-dependent and dialysis-dependent patients. The reason for issuing the CRL was the need for additional clinical studies regarding the safety of roxadustat in both non–dialysis-dependent and dialysis-dependent populations. 5
Akebia Therapeutics received a CRL from the FDA in March 2022 regarding its NDA for use of vadadustat in the treatment of anemia due to CKD. The FDA expressed safety concerns regarding the following: failure to meet noninferiority in major adverse cardiovascular event (MACE) in the non–dialysis-dependent patient population, an increased risk of thromboembolic events (driven by vascular access thrombosis in dialysis patients), and risk of drug-induced liver injury. The CRL recommended new clinical trials to address these issues. 6
Clinical Pharmacology
Daprodustat is an oral HIF-PH inhibitor; it has activity against PH1, PH2, and PH3.1 -4,7 The oxygen sensors (prolyl hydroxylases) help regulate HIF activity through oxygen-dependent proteasomal degradation. When hypoxia is observed, HIF (a transcription factor) stimulates endogenous erythropoietin production by the liver and kidneys and ultimately erythropoiesis.8-10 Daprodustat stabilizes intracellular HIF1α and HIF2α and is thought to induce a transient pseudo-hypoxic state that results in the stimulation of erythropoiesis in CKD patients with anemia.1,2,7 Daprodustat may also influence iron hemostasis through effects on ferritin levels, transferrin saturation, transferrin receptor expression, hepcidin concentrations, and other iron-related proteins.2 -4,7
Daprodustat’s mechanism of action is different from previously used ESAs (eg, epoetin alfa, epoetin beta, rh-epoetin, darbepoetin alfa).10 -12 Daprodustat appears to have 1000-fold or higher selectivity for the 3 prolyl hydroxylase domains (PHD1, PHD2, PHD3), with a preference for PHD1 and PHD3.2,7,10 Roxadustat appears to equally inhibit all 3 enzymes, and vadadustat may have more selective activity against PHD3 and PHD1 than PHD2. 10 There are other HIF-PH inhibitors (eg, desidustat, enarodustat, molidustat, roxadustat, vadadustat) in development.10 -12
Daprodustat’s impact on plasma hemoglobin concentration, serum total iron-binding capacity, and ferritin concentrations is dose related, with higher doses associated with a higher impact on these outcomes.1,13 An increase in endogenous erythropoietin occurred within 6 to 8 hours after administration. With continued administration, weak increases in reticulocyte counts occur between 7 and 15 days, with subsequent increases in red blood cell production. New hemoglobin steady-state levels are reached several weeks after initial administration (approximately 4 weeks with ESA use and 16-20 weeks in those not using ESAs). Improvements in serum transferrin and total iron-binding capacity as well as decreases in serum ferritin, transferrin saturation, and hepcidin were observed over 52 weeks of treatment in adults on dialysis with anemia due to CKD. 1
Single doses of daprodustat 75 and 500 mg did not produce clinically meaningful changes in QT/QTc interval.1,14
Pharmacokinetics
Absolute oral bioavailability is 65%.1,15 Median Tmax following oral administration is 1 to 2 hours. Tmax for the 6 predominant metabolites is slower, with a median Cmax occurring between 2 and 8 hours. 16 Administration with a high-fat/high-calorie meal did not significantly alter daprodustat exposure compared with administration in the fasted state. 1
The terminal half-life of daprodustat was approximately 1 to 4 hours in healthy participants and 7 hours in patients with CKD.1,2,15,16 Daprodustat’s half-life was similar among patients with CKD stage 3 to 4, patients on hemodialysis (when received on dialysis day), and patients on peritoneal dialysis, while in dialysis patients receiving daprodustat on nondialysis days, mean half-life was 18.9 hours.2,16 Daprodustat is mainly metabolized by CYP2C8 (with minor contribution of CYP3A4); therefore, CYP2C8 inhibitors or inducers may alter the plasma concentration of daprodustat when used concomitantly.1,2,15 The percentage excreted unchanged was 0.05% for subjects with normal renal function and 0.04% in those with CKD stage 3 to 4. 16
Following IV administration in healthy adults, volume of distribution is 14.3 L.1 Daprodustat is highly bound to protein (mainly albumin); therefore, the drug is not significantly removed by hemodialysis.1,2
Mean clearance from plasma was 18.9 L/hour, which correlates to blood clearance of 15 L/hour and equates to a hepatic extraction of approximately 18%. Within 7 days of an oral dose of radiolabeled daprodustat, 74% of the radioactivity was recovered in the feces, and 21% in the urine. Approximately 99.5% of the dose was excreted as oxidative metabolites, with the rest accounted for by daprodustat. 1
The steady-state exposure of daprodustat is similar in patients with normal renal function and in those with varying degrees of renal impairment; daprodustat exposure is not significantly impacted by hemodialysis or peritoneal dialysis. The systemic exposure of daprodustat metabolites was higher in patients with stage 3 to 5 CKD compared to those with normal renal function. Exposures of metabolites were higher on nondialysis days compared to dialysis days. 1
Clearance was 0.2 mL/minute in healthy subjects and 0.17 mL/minute in patients with CKD stage 3 to 4. Dialysis clearance was −8.5 mL/minute during hemodialysis and 10.61 mL/minute during peritoneal dialysis. Dialysis clearance of the metabolites was higher in patients on hemodialysis and lower in patients on peritoneal dialysis. 16
Daprodustat Cmax and AUC were increased 2-fold in participants with moderate (Child-Pugh class B) hepatic impairment versus healthy controls; in participants with mild hepatic impairment, Cmax was comparable with controls and AUC was increased 1.5-fold.1,17
Comparative Efficacy
Indication: Anemia in Patients With Chronic Kidney Disease Undergoing Dialysis (FDA-Approved Use)
These studies confirm the results in phase 2 studies.7,18,19
Separate phase 3 studies conducted in Japanese patients undergoing hemodialysis and peritoneal dialysis showed comparable results for daprodustat efficacy and safety.20-22
Studies
Respective baseline characteristics in the daprodustat and ESA groups were as follows: Median patient age was 58 and 59 years; 42.8% and 42.7% were female; 66.9% and 66.5% were White, 15.3% and 15.8% were Black, and 11.8% and 12.3% were Asian; median body mass index (BMI) was 26.8 and 26.8 kg/m2; ESA hyporesponsiveness was 12.3% and 12.2%; percent receiving hemodialysis was 88.5% and 88.6%, and percent receiving peritoneal dialysis was 11.5% and 11.4%; and the duration of dialysis was 0 to less than 2 years in 30.5% and 30.5%, respectively, 2 to less than 5 years in 36% and 35.8%, and 5 or more years in 33.6% and 33.6%. Mean baseline hemoglobin was 10.35 g/dL in the daprodustat group and 10.39 g/dL in the ESA group. About 29% of patients were from the United States.
● Mean change in hemoglobin level from baseline to the primary evaluation period (weeks 28 through 52) was 0.28 g/dL in the daprodustat group and 0.1 g/dL in the ESA group; mean adjusted difference was 0.18 g/dL (95% CI, 0.12-0.24; P < .001), meeting the prespecified noninferiority margin; the prespecified subgroup analysis showed comparable results.
● Occurrence of adjudicated MACE (composite of death from any cause, nonfatal MI, or nonfatal stroke) was 25.2% in the daprodustat group and 26.7% in the ESA group; hazard ratio (HR) was 0.93 (95% CI, 0.81-1.07; P < .001), which was less than the prespecified noninferiority margin.
● Incidence of MACE (on treatment analysis) was 25.2% in the daprodustat group and 26.7% in the ESA group.
● Superiority testing results for the first occurrence of various secondary cardiovascular outcomes were not significant: MACE (HR, 0.93; 95% CI, 0.81-1.07), MACE or a thromboembolic event (HR, 0.88; 95% CI, 0.78-1), and MACE or hospitalization for heart failure (HR, 0.97; 95% CI, 0.85-1.11).
● Adjusted mean monthly IV iron dose from baseline to week 52 was 90.8 mg in the daprodustat group and 99.9 mg in the ESA group; mean difference was −9.1 mg (95% CI, −18.4 to 0.2).
● Incidence of all-cause mortality (primary safety analysis) was 16.4% in the daprodustat group and 15.8% in the ESA group.
Rescue therapy was required by 3.6% in both groups; the percentage that required at least one red blood cell or whole blood transfusion during the treatment period was 15.7% in the daprodustat group and 18.3% in the ESA group (HR, 0.86; 95% CI, 0.72-1.02). Median duration of follow-up for evaluation of cardiovascular events was 2.5 years and provided 7,028 total person-years of follow-up.
In the daprodustat ITT cohort, median patient age was 52 years; 61% were male; and 70% were White, 17% were Asian, and 10% were Black or African American. In the darbepoetin alfa ITT cohort, median patient age was 56 years; 63% were male; and 69% were White, 20% were Asian, and 8% were Black or African American. The majority of patients were undergoing hemolysis (80% and 81%, respectively), with dialysis planned in 69% in both groups; baseline mean hemoglobin was 9.5 g/dL in both groups; median transferrin saturation was 28% and 30%, respectively; ferritin was 365 and 373 ng/mL, respectively; oral iron was used by 16% and 14% and IV iron was used by 67% and 70%, respectively (median standardized IV iron dose at baseline was 87 and 130 mg/month, respectively).
● Mean hemoglobin concentration during the evaluation period was 10.5 g/dL in the daprodustat arm and 10.6 g/dL in the darbepoetin alfa arm. Mean hemoglobin concentration for both arms remained in the analysis range of 10 to 11.5 g/dL. Adjusted mean change from baseline in hemoglobin level was 1.02 and 1.12 g/dL, respectively; treatment difference was −0.1 g/dL (95% CI, −0.34 to 0.14), which met noninferiority criteria.
●
● Reduction in mean monthly IV iron dose from baseline to week 52 was similar for the 2 treatment groups (adjusted mean treatment difference was 19.4 mg/month [95% CI, −11 to 49.9]); mean monthly IV iron use was 142 mg in the daprodustat group and 137 mg in the darbepoetin alfa group.
● Adjudicated MACE-type adverse reactions: MACE occurred in 12% of the daprodustat group and 10% of the darbepoetin alfa group; all-cause mortality occurred in 9% and 6%, respectively. The incidence rate per 100 patient-years was 11.65 and 9.24; absolute rate difference per 100 patient-years was 2.41 (−4.61 to 9.43).
Subgroup analysis (based on hemodialysis and peritoneal dialysis and planned and unplanned dialysis start) for the primary hemoglobin outcome showed similar results. Noninferiority of daprodustat compared with darbepoetin alfa was declared if the lower bound of the 2-sided 95% CI of the difference in hemoglobin concentration between treatments exceeded −0.75 g/dL.
The efficacy analysis was evaluated in the ITT cohort, with missing hemoglobin values being imputed using multiple imputation under a “missing at random” assumption. Rescue therapy was only required by 3% of patients in both groups.
● Treatment difference in mean change in hemoglobin during the evaluation period (weeks 28-52) was −0.05 g/dL (95% CI, −0.21 to 0.1), which met criteria for noninferiority. Mean hemoglobin level during the evaluation period was 10.45 g/dL in the daprodustat group and 10.41 g/dL in the epoetin alfa group.
● Average monthly IV iron requirements were not statistically significantly different between the 2 groups: 98.11 mg in the daprodustat group and 106.23 mg in the epoetin alfa group; treatment difference was −8.12 mg (95% CI, −45.66 to 29.41).
● Proportion of responders (in hemoglobin analysis range 10-11.5 g/dL) over evaluation period: 80% of the daprodustat group were classified as responders compared with 63.6% of the epoetin alfa group; treatment difference was 16.5% (1-sided nominal P < .001 after adjustment for region).
● Fewer blood pressure elevations occurred in the daprodustat group than in the epoetin alfa group (1-sided nominal P = .009), and the overall effect on blood pressure was similar with both drugs.
The manufacturer is required to conduct 2 observational studies comparing daprodustat and an ESA comparator arm. A prospective observational study (at least 5-year follow-up) in the United States is being conducted to characterize the long-term safety (eg, MACE, hospitalization for heart failure, serious GI bleeds, eye disorders, hepatic injury) of daprodustat in adults with dialysis-dependent CKD treated with the approved dosing regimen; an interim report is due in July 2028 and the study must be completed by July 2031, with the final report submitted by July 2032. An observational study (at least 5-year follow-up) is being conducted to assess the risk of malignancy in adults with dialysis-dependent CKD; the study must be completed by October 2031, with the final report submitted by October 2032. 28
Indication: Anemia in Patients With Chronic Kidney Disease Not Undergoing Dialysis (Non–FDA-Approved Use)
These studies confirm results in phase 2 studies.7,29
A separate phase 3 study conducted in Japanese patients not on dialysis showed comparable results regarding daprodustat efficacy and safety compared with epoetin beta pegol. 30
Network meta-analyses of the efficacy and safety of HIF-PH inhibitors, epoetin, and darbepoetin for anemia of CKD in patients not undergoing dialysis determined there was no difference between the various drugs regarding elevation of hemoglobin levels; compared with placebo, there was no significant association between the various drugs and all-cause mortality.11,12,31
Studies
Median patient age was 67 years in both the daprodustat and darbepoetin alfa groups; 56.9% and 55.3%, respectively, were female; 56.7% and 54.5% were White, 27.1% and 27.8% were Asian, and 9.4% and 9.6% were Black; median BMI was 26.9 and 26.6 kg/m2; proportion not using an ESA was 53.2% and 53.3%; and 82.2% and 80.8% had stage 4 or 5 CKD. Mean baseline hemoglobulin was 9.9 g/dL in the daprodustat group and 9.8 g/dL in the darbepoetin alfa group. About 25% of patients were from the United States.
● Mean change in hemoglobin level from baseline to the primary evaluation period (weeks 28 through 52) was 0.74 g/dL in the daprodustat group and 0.66 g/dL in the darbepoetin alfa group; mean adjusted difference was 0.08 g/dL (95% CI, 0.03-0.13; P < .001), which met the prespecified noninferiority margin; the prespecified subgroup analysis showed comparable results.
● Proportion of patients with occurrence of MACE (composite of death from any cause, nonfatal MI, or nonfatal stroke) was 19.5% in the daprodustat group and 19.2% in the darbepoetin alfa group; HR was 1.03 (95% CI, 0.89-1.19; P = .005), which was less than the prespecified noninferiority margin.
● Incidence of MACE (on treatment analysis, which censored data on patients at 28 days after the date of the last dose) was 14.1% in the daprodustat group and 10.5% in the darbepoetin alfa group; HR was 1.4 (95% CI, 1.17-1.68). This analysis did not consider the different dosing intervals between the 2 treatment groups or the impact of the observation periods. Therefore, post hoc analyses were conducted to explore the effect of the different dosing frequencies; results were more consistent with the results of the primary analysis.
● Superiority testing results for the first occurrence of various secondary cardiovascular outcomes were not significant: MACE (HR, 1.03; 95% CI, 0.89-1.19), MACE or a thromboembolic event (HR, 1.06; 95% CI, 0.93-1.22), MACE or hospitalization for heart failure (HR, 1.09; 95% CI, 0.95-1.24), and CKD progression (HR, 0.98; 95% CI, 0.84-1.13).
● Incidence of all-cause mortality was similar between treatment groups; HR was 1.03 (95% CI, 0.87-1.2).
Adherence to the assigned therapy was 97% in the daprodustat group and 98% in the darbepoetin alfa group. Rescue therapy was required by 2% of the daprodustat group and 3.3% of the darbepoetin alfa group; the percentage that required at least 1 red blood cell or whole blood transfusion during the treatment period was 12.8% in the daprodustat group and 13.5% in the darbepoetin alfa group (HR, 0.96; 95% CI, 0.81-1.14). Discontinuation of treatment for reasons other than death occurred in 29.5% of the daprodustat group and 28.9% of the darbepoetin alfa group. Median duration of follow-up for evaluation of cardiovascular events was 1.9 years, which provided 7,210 total person-years of follow-up.
Mean patient age was 65.9 years; 57.7% were female; and 63.8% were White, 14.8% were Black or African American, and 11.1% were American Indian or Alaska Native.
● Mean change in hemoglobin between baseline and the evaluation period (mean value over week 24-28) was 1.58 g/dL with daprodustat and 0.19 g/dL with placebo; adjusted mean difference in hemoglobin was 1.4 g/dL (95% CI, 1.23-1.56; P < .001).
● Proportion of patients with a 1 g/dL or more increase in hemoglobin was 77% with daprodustat and 18% with placebo (P < .001); estimated difference in response rate was 0.56 (95% CI, 0.49-0.63).
● Mean change in SF-36 vitality (fatigue) score from baseline to week 28 was 7.3 points with daprodustat and 1.9 points with placebo (P < .001); mean difference was 5.36 (95% CI, 2.17-8.56).
● Percentage of patients with hemoglobin response (achievement of hemoglobin of 11-12 g/dL) during the evaluation period (weeks 24-28) was 52% with daprodustat and 8% with placebo; estimated value for difference in response rate was 0.45 (95% CI, 0.37-0.52).
● Proportion of patients who were SF-36 vitality responders (6-point or more increase) was 58% with daprodustat and 40% with placebo; treatment difference was 13% (95% CI, 4% to 22%).
● Blood pressure elevations occurred more often with daprodustat (32%) than placebo (26%) (P = .07); however, the overall effect of daprodustat therapy on blood pressure was similar to placebo.
Contraindications, Warnings, and Precautions
Contraindications
Daprodustat is contraindicated in patients requiring treatment with strong CYP2C8 inhibitors (eg, gemfibrozil). 1
Daprodustat is contraindicated in patients with uncontrolled hypertension. 1
Though not stated in the daprodustat product labeling, a potential contraindication to use is hypersensitivity reactions to daprodustat or to any of the product ingredients (ie, colloidal silicon dioxide, croscarmellose sodium, hypromellose, magnesium stearate, mannitol, microcrystalline cellulose, hypromellose, iron oxide black, iron oxide red, iron oxide yellow, polyethylene glycol, titanium dioxide). 1
Warnings and Precautions
Daprodustat labeling includes a boxed warning regarding the risk of death, MI, stroke, venous thromboembolism, and thrombosis of vascular access during treatment with daprodustat. The risk of potentially fatal thrombotic vascular events, including MACE, is increased during treatment with daprodustat. Daprodustat use should be avoided in patients with a history of MI, cerebrovascular events, or acute coronary syndrome within the past 3 months. 1
A rise in hemoglobin greater than 1 g/dL over 2 weeks may contribute to these risks. Also, a target hemoglobin level greater than 11 g/dL may increase the risk of death and arterial venous thrombotic events, a risk that is also associated with the use of ESAs. It is unknown if adjustments to the hemoglobin target level, dose, or dosing strategy can decrease these risks. Therefore, the lowest dose of daprodustat sufficient to reduce the need for red blood cell transfusions should be prescribed, and adherence to dosing and hemoglobin monitoring is recommended to avoid excessive erythropoiesis. 1
Risk of hospitalization for heart failure may be higher with daprodustat therapy. In the ASCEND-D trial, hospitalization for heart failure occurred in 7.5% (3.3 per 100 person-years) of patients on dialysis receiving daprodustat and 6.8% (3 per 100 person-years) of patients receiving recombinant human erythropoietin (rhEPO). Patients with preexisting heart failure were at increased risk of hospitalization for heart failure with daprodustat (14.5%; 6.8 per 100 person-years) compared with rhEPO (11.3%; 5.1 per 100 person-years). 1
Uncontrolled hypertension is a contraindication to daprodustat therapy. In the ASCEND-D trial, worsening of hypertension occurred in 24% (12 per 100 person-years) of patients receiving daprodustat and 24% (12 per 100 person-years) of patients receiving rhEPO. Serious worsening of hypertension occurred in 3.1% of patients receiving daprodustat and 3.1% of patients receiving rhEPO. Cases of hypertensive crisis, including hypertensive encephalopathy and seizures, have also been reported. Periodically monitor blood pressure and adjust or initiate antihypertensive therapy as needed. 1
GI erosion may occur during daprodustat therapy. In the ASCEND-D trial, gastric or esophageal erosions occurred in 5.7% (2.5 per 100 person-years) of patients receiving daprodustat and 6.6% (2.9 per 100 person-years) of rhEPO-treated patients. Serious erosions, including GI bleeding and the need for red blood cell transfusions, were reported in 3.6% and 3.1% of patients receiving daprodustat and rhEPO, respectively. Consider this risk particularly in patients at increased risk for GI erosions, such as those with a history of GI erosion, peptic ulcer disease, use of concomitant medications that increase the risk of GI erosion, and current tobacco and alcohol use. 1
The safety of daprodustat has not been sufficiently established for the treatment of anemia due to CKD in adults not on dialysis, and this use is not FDA approved. In ASCEND-ND, an increased risk of cardiovascular mortality, stroke, thromboembolism, serious acute kidney injury, hospitalization for heart failure, and serious GI erosions was observed in patients treated with daprodustat compared with rhEPO. 1
The safety of daprodustat in patients with malignancy has not been established. Based on its mechanism of action (increased HIF-1 levels), daprodustat may have an unfavorable effect on cancer growth and therefore should be avoided in patients with active malignancies. Malignancies were observed in 4.4% (1.9 per 100 person-years) of patients treated with daprodustat and 5.2% (2.3 per 100 person-years) of patients treated with rhEPO. 1
There are no adequate and well-controlled studies of daprodustat use during pregnancy. CKD in pregnancy increases the risk for maternal hypertension, preeclampsia, miscarriage, stillbirth, preterm delivery, low–birth weight infants, and polyhydramnios. Daprodustat in animal studies was associated with adverse fetal outcomes. Pregnant patients should be advised of the potential risk to the fetus. 1 A prospective and retrospective evaluation of data from patients exposed to daprodustat during pregnancy is required to assess the risk of pregnancy and maternal complications, adverse effects on the developing fetus and neonate, and adverse effects on the infant. An interim report is due by September 2027 and the study must be completed by August 2029, with the final report submitted by September 2030. 28
Caution should be used when administering daprodustat during breastfeeding. No studies have been conducted to assess the presence of daprodustat in human milk or its effects on breastfeeding infants or milk production. Daprodustat is present in the milk of lactating rats and, therefore, would likely be present in human milk. Because of the potential for serious adverse reactions (eg, thrombotic vascular events), patients should be advised not to breastfeed during daprodustat therapy and for 1 week after the final dose. 1
Safety and effectiveness of daprodustat have not been established in pediatric patients. 1 A study is required to evaluate the pharmacokinetics, pharmacodynamics, and safety of daprodustat for the treatment of anemia associated with CKD in children and adolescents aged 3 months to younger than 18 years requiring dialysis. The study must be completed by August 2029, with the final report submitted by February 2030. 28
No overall differences in safety and efficacy were observed in patients 65 years and older and younger adults treated with daprodustat. 1
Adverse Reactions
The most common adverse reactions associated with daprodustat (incidence at least 10%) are hypertension, thrombotic vascular events, and abdominal pain. 1
See Table 1 for adverse reactions reported in patients with anemia undergoing dialysis (ASCEND-D study). 4 See Table 2 for adverse reactions reported in patients with anemia not undergoing dialysis (ASCEND-ND study). 3 See Table 3 for a summary of key adverse effects highlighted in the prescribing information. 1
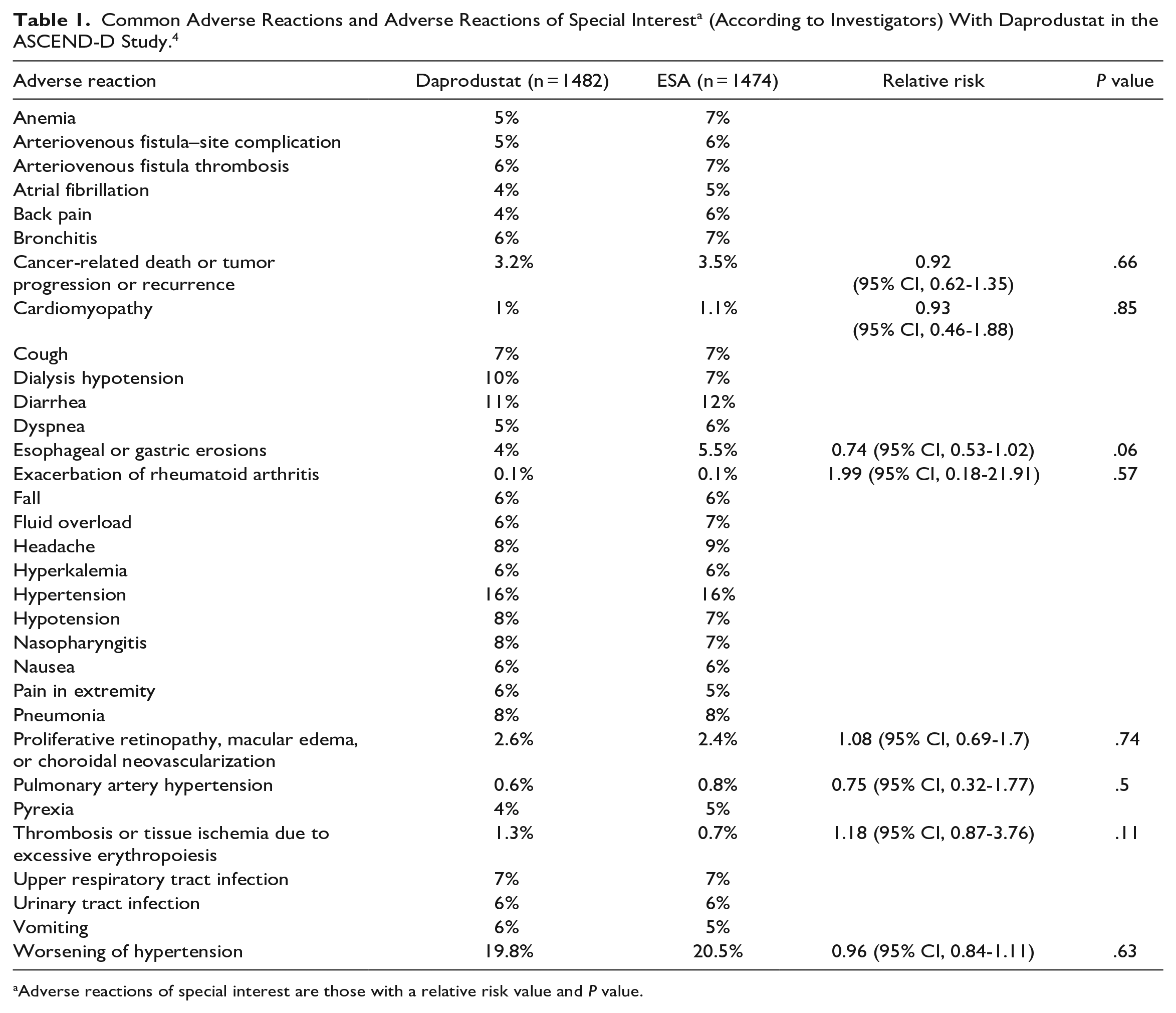
Adverse reactions of special interest are those with a relative risk value and P value.
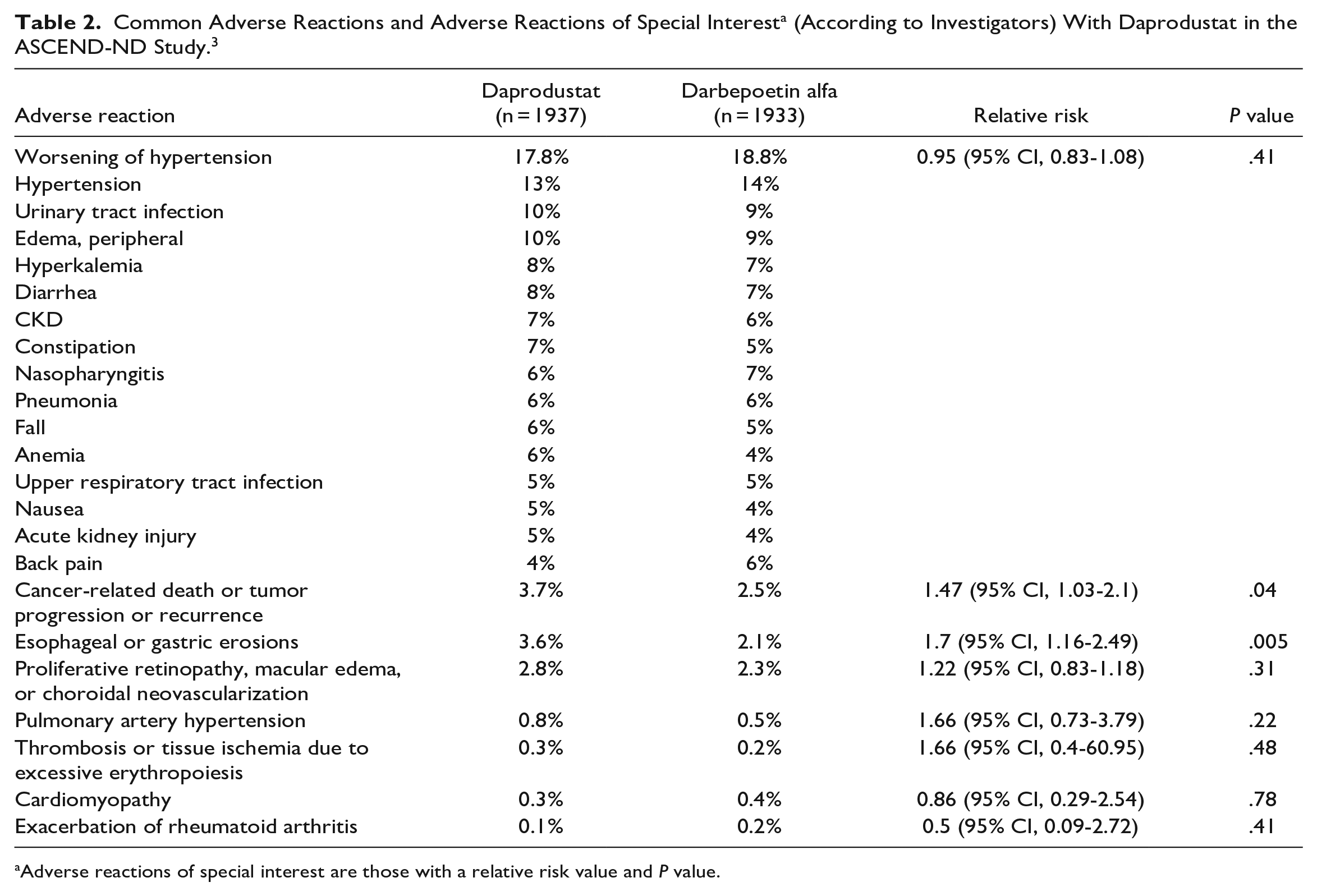
Adverse reactions of special interest are those with a relative risk value and P value.
Adverse Reactions Highlighted in the Daprodustat Prescribing Information (ASCEND-D Study). 1
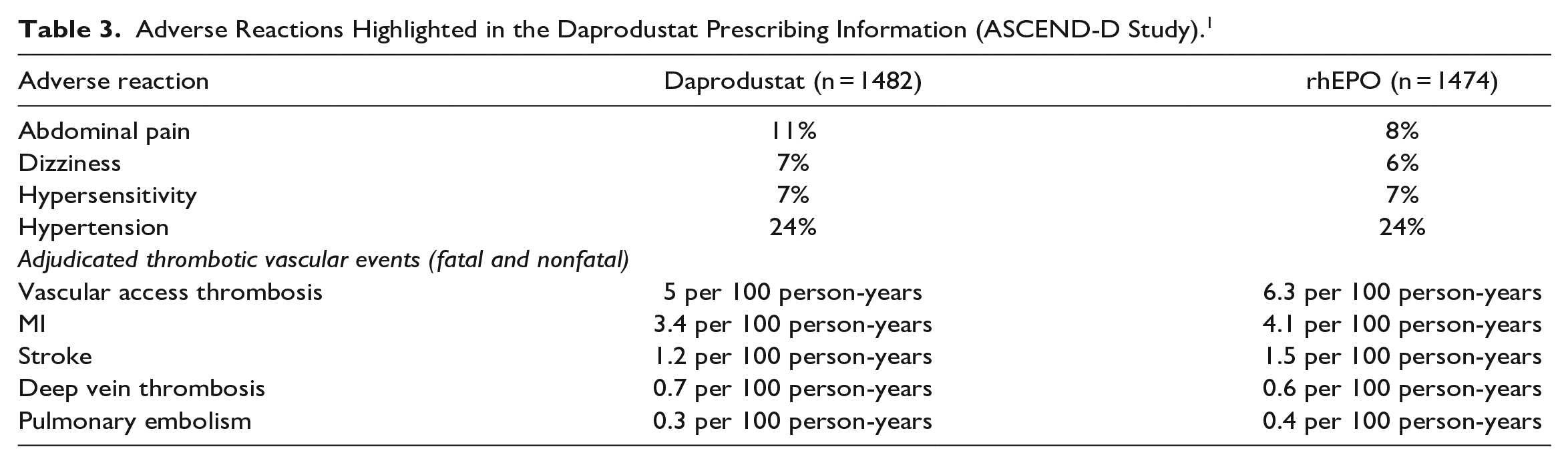
The manufacturer is required to complete a pharmacovigilance analysis of all serious adverse events of GI erosion and hypertensive crisis. The periodic adverse drug experience report is due quarterly during the first 3 years post approval and annually thereafter, through the fifth year following the initial US approval date. The manufacturer must also submit daprodustat utilization reports, including use in non–dialysis-dependent patients and in females of reproductive potential (ie, 15-50 years of age), calculated cumulatively from the initial approval date; utilization reports must be submitted annually through the fifth year following the initial US approval date. 28
Drug Interactions
Daprodustat is a CYP2C8 substrate; inhibitors (eg, clopidogrel, trimethoprim) or inducers (eg, rifampicin) of this enzyme may alter daprodustat plasma concentration.1,2,36
Concomitant administration of moderate CYP2C8 inhibitors (eg, clopidogrel) increases daprodustat exposure. The starting daprodustat dose should be reduced by half, except in patients whose starting dose is already 1 mg, when initiating treatment in patients receiving clopidogrel or a moderate CYP2C8 inhibitor. Hemoglobin should be monitored and dose adjusted when initiating or stopping therapy with clopidogrel or a moderate CYP2C8 inhibitor during treatment with daprodustat. 1
Concomitant use of a CYP2C8 inducer (eg, rifampin) may decrease daprodustat exposure; hemoglobin levels need to be monitored and the dose of daprodustat adjusted when initiating or stopping therapy with CYP2C8 inducers. 1
Recommended Monitoring
Hemoglobulin levels should be obtained at baseline and periodically throughout therapy. 1
Blood pressure should be monitored throughout therapy. 1
Patients should be advised to report any signs or symptoms of MACE, heart failure, or gastric or esophageal erosions and GI bleeding. 1
Dosing
Prior to starting daprodustat treatment, the cause of the anemia needs to be determined (eg, vitamin deficiency, metabolic or chronic inflammatory conditions, bleeding) and corrected, and iron status needs to be established. If serum ferritin is less than 100 µg/mL or serum transferrin saturation is less than 20%, supplemental iron therapy should be initiated; the majority of patients require supplemental iron during the course of therapy. Liver function tests should also be obtained (eg, ALT, AST, alkaline phosphatase, total bilirubin). If the patient develops signs or symptoms consistent with liver disease during daprodustat treatment, the tests should be repeated. 1
The dose of daprodustat needs to be individualized to reduce the need for red blood cell transfusions and to maintain a hemoglobin level of less than or equal to 11 g/dL.1
The starting dose of daprodustat for adults with anemia due to CKD receiving dialysis for at least 4 months is based on hemoglobin level and previous ESA use (see Tables 4 and 5). Dose modifications are needed for patients receiving concomitant treatment with a moderate CYP2C8 inhibitor or who have moderate hepatic impairment. 1
Starting Daprodustat Dose for Adults on Dialysis Not Receiving an ESA. 1

Starting Daprodustat Dose for Adults on Dialysis Switching From an ESA. 1

Hemoglobin levels should be obtained every 2 weeks after initiation of treatment and after each dose adjustment for the first month and then every 4 weeks thereafter. Dosage adjustments should be based on the hemoglobin rate of rise, rate of decline, and hemoglobin variability, but increases in dose should be no more frequent than once every 4 weeks. If a dose adjustment is necessary, the dose should be increased or decreased by one dose level at a time (1, 2, 4, 6, 8, 12, 16, and 24 mg). The maximum recommended daily dose of daprodustat is 24 mg. The dose of daprodustat should be decreased if hemoglobin increases rapidly (eg, greater than 1 g/dL over 2 weeks or greater than 2 g/dL over 4 weeks) or if hemoglobin exceeds 11 g/dL. If hemoglobin exceeds 12 g/dL, interrupt treatment with daprodustat. Treatment can be restarted at one dose level lower when hemoglobin returns to within the target range. If no clinically meaningful increase in hemoglobin level is achieved within 24 weeks, treatment with daprodustat should not be continued; instead, alternative explanations for an inadequate response should be sought and treated before restarting daprodustat treatment. 1
Patients with moderate hepatic impairment (Child-Pugh class B) should receive a lower daily dose of daprodustat; the starting dose should be reduced by 50% except in patients whose starting dose is already 1 mg. Daprodustat therapy is not recommended in patients with severe hepatic impairment (Child-Pugh class C). 1
Concomitant treatment with moderate CYP2C8 inhibitors or clopidogrel requires a 50% reduction in the starting dose of daprodustat, except for patients whose starting dose is already 1 mg. If the CYP2C8 inhibitor or clopidogrel therapy is discontinued, the hemoglobin level should be monitored and the dose of daprodustat adjusted accordingly. 1
Daprodustat tablets should be swallowed whole once daily; do not cut, crush, or chew. 1
Daprodustat can be administered without regard to timing or type of dialysis. 1
If a dose is missed, it should be taken as soon as possible, unless it is the same day as the next dose. In this case, the missed dose should be skipped, and the next dose should be taken at the usual time; do not double a dose to make up for a missed dose. 1
Product Availability and Storage
The NDA for daprodustat (for treatment of anemia associated with CKD in adults receiving dialysis) was accepted by the FDA in April 2022, with a Prescription Drug User Fee Act date of February 1, 2023. 37 Daprodustat was approved by the FDA on February 1, 2023. 28
Daprodustat is available as 1, 2, 4, 6, and 8 mg tablets in bottles of 30. 1
The tablets should be stored at 20°C to 25°C (68°F-77°F); excursions are permitted between 15°C and 30°C (59°F and 86°F). 1
Drug Safety/REMS
No REMS is required for daprodustat. 1
Conclusion
Daprodustat is a HIF-PH inhibitor FDA approved for the treatment of anemia associated with CKD in adults receiving dialysis for at least 4 months. Its mechanism of action is different from the ESAs, which may make it a useful alternative in patients unable to tolerate ESAs or with an inadequate response to an ESA plus adequate iron supplementation. Based on results from phase 3 clinical trials, daprodustat is capable of improving and maintaining hemoglobin concentrations and other hematopoietic indicators in both non–dialysis-dependent and dialysis-dependent patients; however, FDA approval is only for use in the treatment of anemia due to CKD in adults receiving dialysis.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.