
Abstract
In this study, we synthesized styrene–methyl methacrylate copolymer particles by suspension polymerization process to study the foaming dynamics via visual batch foaming apparatus. The weight ratio of styrene–methyl methacrylate was 53–47. The foaming system consisted of a self-sealing observation cell equipped with two glass windows and a stereo microscope attached with a high-speed digital camera as well as a pressure and temperature controller. The required pentane pressure was supplied by heating a small pentane flask. The synthesized copolymer particles impregnated by pentane and then the foaming dynamics were recorded after a rapid pressure release. The effect of different foaming conditions, such as temperature, impregnation time, and impregnation pressure on the expansion ratio of styrene–methyl methacrylate copolymer was investigated. It was concluded that impregnation pressure, time, and temperature have different effects on the foaming ratio at impregnation pressures lower and higher than 4 bar.
Introduction
The expandable pattern casting (EPC) or the lost foam casting process is an economic and most widely used method to produce complex metal parts, in which the liquid metal displaced the refractory-coated expandable foam patterns by thermal degradation. The EPC technology has different advantages compared to the conventional casting methods in which the cast is made of expanded polystyrene (EPS) in aluminum casting or expanded styrene–methyl methacrylate (St–MMA) copolymer in iron casting. 1 Using styrenic/acrylic copolymers foams instead of EPS can improve the quality of the produced foam and St–MMA copolymer is of particular importance when considered as an alternative for EPS due to its unique chemical properties. 2 Suspension polymerization is commonly used for producing a wide variety of commercially important polymers such as polystyrene and its copolymers, polymethyl methacrylate (PMMA), polyvinyl acetate, and polyvinyl chloride. It is carried out by suspending the monomer (discontinuous phase) as droplets (50–500 µm in diameter) in water (continuous phase).
Thermoplastic foamed products have become very popular in recent years. The porous structure is achieved through the expansion of a blowing agent dissolved in a thermoplastic by a batch or continuous foam process. 3 – 8 A typical foaming process has three stages in which the first step is polymer/blowing agent dissolution, the second one the cell nucleation, and the last one the cell growth. 9 The first step is achieved by saturating a polymer with a blowing agent, typically above its melting or glass transition temperature. This step is called the impregnation period in which after that, with a rapid pressure drop or temperature increment, thermodynamic instability is introduced in the polymer/gas mixture and cell nucleation is started. Following this, the gas diffuses from the polymer matrix into the cells and cell growth goes ahead. The final properties of polymeric foams in batch systems are affected by the type and amount of the dissolved blowing agent, as well as the foaming parameters such as temperature, impregnation pressure, time, and pressure release rate. The common blowing agents for polymeric foams were chlorofluorocarbons, but their application has been limited due to the ozone depletion concerns. Volatile organic compounds like pentane are sometimes used as physical blowing agents, but they are often flammable. A suitable nucleating agent should be used to produce desirable foams with a narrow cell-size distribution.
Visual studies of the foaming dynamics have been conducted by different researchers. Gent and Tompkins 10 developed a visual observation apparatus to observe foaming of elastomer/CO2 and reported the change in bubble radius with time. Villamizar and Han 11 used visual observation of structural foaming in injection molding of a mixture of polyethylene and chemical blowing agent using a rectangular mold cavity with glass windows on both sides. Yoo and Han 12 designed a visual observation apparatus and compared the experimental data and the numerical modeling of foaming behavior. Actually, a few foaming experiments have focused on in situ visual foaming dynamics. Taki et al. 13 used visual observation of batch and continuous foaming using CO2 as a physical foaming agent to track the bubble nucleation and growth behaviors. Leung et al. 14 used the batch visualization data obtained from an experimental system to investigate the bubble growth dynamics and verified the results with a theoretical model. Salejova and Kosek 15 studied the initial foaming stage through a batch visual system and discussed about the effects of different parameters on the foaming dynamics of polystyrene. Recently, Taki 16 analyzed the effect of pressure release rate on the bubble nucleation and growth via batch foaming experiments, in which the flux of the blowing agent from the polymer matrix to the bubble increased with increasing the pressure release rate. Different researchers have previously analyzed bubble-growth phenomena experimentally in different systems. 17 – 21
In our previous work, suspension copolymerization of St–MMA copolymer particles was conducted and the effects of different synthesis parameters investigated. 2 Subsequently, the non-isothermal degradation kinetics of synthesized St–MMA copolymer foams was investigated under nitrogen and oxygen atmospheres.22,23 But, there are no appreciable studies about the foaming dynamics of this copolymer. In this study, we synthesized St–MMA copolymer particles by suspension polymerization process to investigate the foaming dynamics via visual batch foaming apparatus to examine the effect of different foaming conditions, such as temperature, impregnation pressure, and time, on the foaming ratio of the synthesized copolymer.
Experimental
Materials
MMA and St monomers (Merck, Germany) were washed twice with 5 wt% aqueous solution of sodium hydroxide followed by washing twice with distilled water to eliminate the inhibitor. Benzoyl peroxide (BPO, active content 75%, water 25%) as radical initiator with t1/2 = 75 min at 90°C, poly(vinyl alcohol) (PVA) with a degree of hydrolysis 72.5% and molecular weight (MW) 72,000, hydroxyethyl cellulose (HEC), and poly(vinyl pyrrolidone) (PVP, 360,000 g/mol) as drop stabilizers and potassium persulfate (K2S2O8) as an aqueous phase initiator were supplied by Merck. Tricalcium phosphate (TCP) and sodium dodecyl benzene sulfonate (DBSNa) as suspending agents were supplied by Fluka and were used as received. n-Pentane (Merck, Germany) which was used as the blowing agent and polyethylene wax (PW, 1% of monomer mixture weight) nucleating agent as a cell structure modifying aid were supplied by Tabriz Petrochemical Company, Iran. Silicone oil (Therm 420) was supplied by Lauda and was used as received for heating the polymerization reactor.
Suspension copolymerization
Suspension copolymerization of St–MMA was carried out in a laboratory scale reactor. A 1 -L stainless steel reactor was equipped with chilled water, silicone oil jacket, a nitrogen inlet and outlet for purging, and one baffle. The system was equipped with a proportional–integral–derivative controller to control the temperature inside the reactor by adjusting the temperature of the circulating silicone oil. 2 The St–MMA copolymer particles were synthesized by batch suspension copolymerization. The required amounts of PVA, TCP, PVP, HEC, DBSNa, and K2S2O8 were fed into the reactor containing a pre-weighed amount of distilled deionized water. The mixture was stirred at ambient temperature under a moderate stirring rate (280 rpm) for at least a few hours to produce a homogeneous mixture. A monomer emulsion was made by adding a mixture of St and MMA monomers, free radical initiator (BPO), and cell structure improver (PW). Low MW PW is normally used in industry for cell-size control. Above its melting point, PW will dissolve in the St monomer. With growing conversion, phase inversion occurs and the domain size of polyethylene is set after the cooling step. The PW domains operate as gathering sites for the blowing agent, which are able to form bubbles. So, one of the most important factors in the synthesis of expandable copolymer beads is nucleation agent concentration, which has a significant effect on the expansion properties of these beads. 2
After the mixture was being stirred for 30 min, the content of the monomer emulsion tank was transferred to the reactor. After all the reaction mixtures were added and held for 30 min, the reactor was heated to the reaction temperature (80°C) at a rate of 0.5°C/min and held for 7 hours. After completion of the reaction, the reactor was cooled to 35°C at a rate of 5°C/min. The reaction mixture was transferred out to a batch tank without washing the residual material into the tank. The reaction mixture containing the St–MMA copolymer was acidified to a pH of 1.6–2.0 and after holding for 15 min, the residue was washed with water, dried, and sieved. 2
MW and MW distribution
MW and MW distribution (MWD) which control the processability of the polymer directly affect the expansion and mechanical properties of the final polymer foams. Commonly, size-exclusion chromatography is used for MWD measurements. Meanwhile, this method is insensitive for measuring high MWs which strongly affect polymer processability. Therefore, in this study, the viscoelastic and rheological measurements were used to provide information about MW and MWD of St–MMA expandable copolymer. The complex modulus, G*(ω), as well as the storage and loss moduli, G′(ω) and G″(ω), respectively, are inputs for derivation of MWD. The first step in transformation from G(ω) to MW fraction, w(M), is the computation of the linear relaxation time spectrum, H(τ). This function can be appreciated from its relationship to the linear relaxation modulus, G(t). Calculation of H(τ) from either G*(ω) or G(t) is not straightforward, but once this is done, H(τ) can be used to generate w(M). An approximation method based on the double reptation rule is used as given below:
24
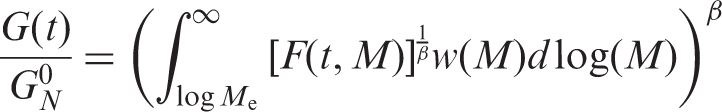
MWD was determined from melt rheometry using a Physica MCR 301 (Anton Paar, Austria) strain controlled rheometer.
Copolymer analysis
Particle composition was determined by (1H-NMR; proton nuclear magnetic resonance). The particles were dissolved in deuterated chloroform, CDCl3; the spectra of the samples were obtained using a Brüker (Adavance DPX) NMR spectrometer working at 500 MHz. The molar percentage of monomers incorporated to the particles was determined from the peak of OCH3 group (at 3.6 ppm), and the peaks correlated to the aromatic ring (between 6.5 and 7.5 ppm). The peaks that correlated to the CH2 and CH groups were used to verify the integration error. The copolymer composition from the1H-NMR spectrum was evaluated using the following equation:
28
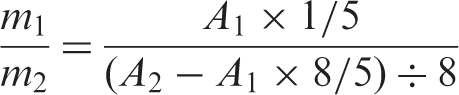
Differential scanning calorimetry (DSC) thermograms were recorded using a Netzsch-DSC200 F3 Maia for glass transition temperature (Tg) determination. All experiments were carried out in an inert dynamic atmosphere of high-purity nitrogen set at a flow rate of 50 mL/min. Five milligrams of the synthesized beads was taken in a standard 40 µL aluminum crucible and temperature scanning was conducted from room temperature to 180°C at a heating rate of 10°C/min.
Foaming procedure
The visual batch foaming system consisted of the self-sealing observation cell equipped with two glass windows, a Bell stereo microscope attached with a Bell high-speed digital camera, as well as a pressure and temperature digital controller. The St–MMA copolymer particles were placed into the observation chamber on the metal grid between the two glass windows and are illuminated by a light source either from the top or the bottom and heated electrically. The digital camera was connected to the computer equipped with a digital image processing software and all changes in the particle size were detected. The setting of the microscope was kept constant during the experiments. The synthesized copolymer particles impregnated by pentane and then foaming dynamics were recorded as the sequence of images of expanding particles located in the pressure cell. The required pentane pressure was supplied by heating a small pentane flask.
All the experiments were run at constant temperature of the observation cell (between 85°C and 105°C). The St–MMA copolymer particle was inserted into the observation cell and heated up to the required temperature. Then, the sorption of pentane in the particles was conducted at different pressures of pentane vapor for a specified period of impregnation time (timpreg) and finally the pressure was quickly released and the foaming procedure of the particles was recorded at regular time intervals of 10 s.
The foam cell structure was studied using a scanning electron microscope (SEM, VEGA-TESCAN). The foam samples were gold-coated before SEM examination.
Results and discussion
Copolymer composition and structure
The1H-NMR spectrum of the St–MMA copolymer is shown in Figure 1. The copolymer composition from the spectrum was evaluated using Equation (2) as an St/MMA ratio of 53/47. The peaks at 7.1 and 3.6 ppm correspond to –C6H5 and –OCH3 groups, respectively. The peak at 2.8 ppm, absent in the1H-NMR spectra of both relevant homopolymers,
29
is observed in the copolymer spectrum. This has been assigned to the –OCH3 group of MMA bonded to the St sequence.
30
Opresnik et al.
31
also showed that this peak could be much more clearly seen in the1H-NMR spectra of a St–MMA random copolymer rather than that of a St–MMA block copolymer.
The1H-NMR spectrum of the St–MMA copolymer.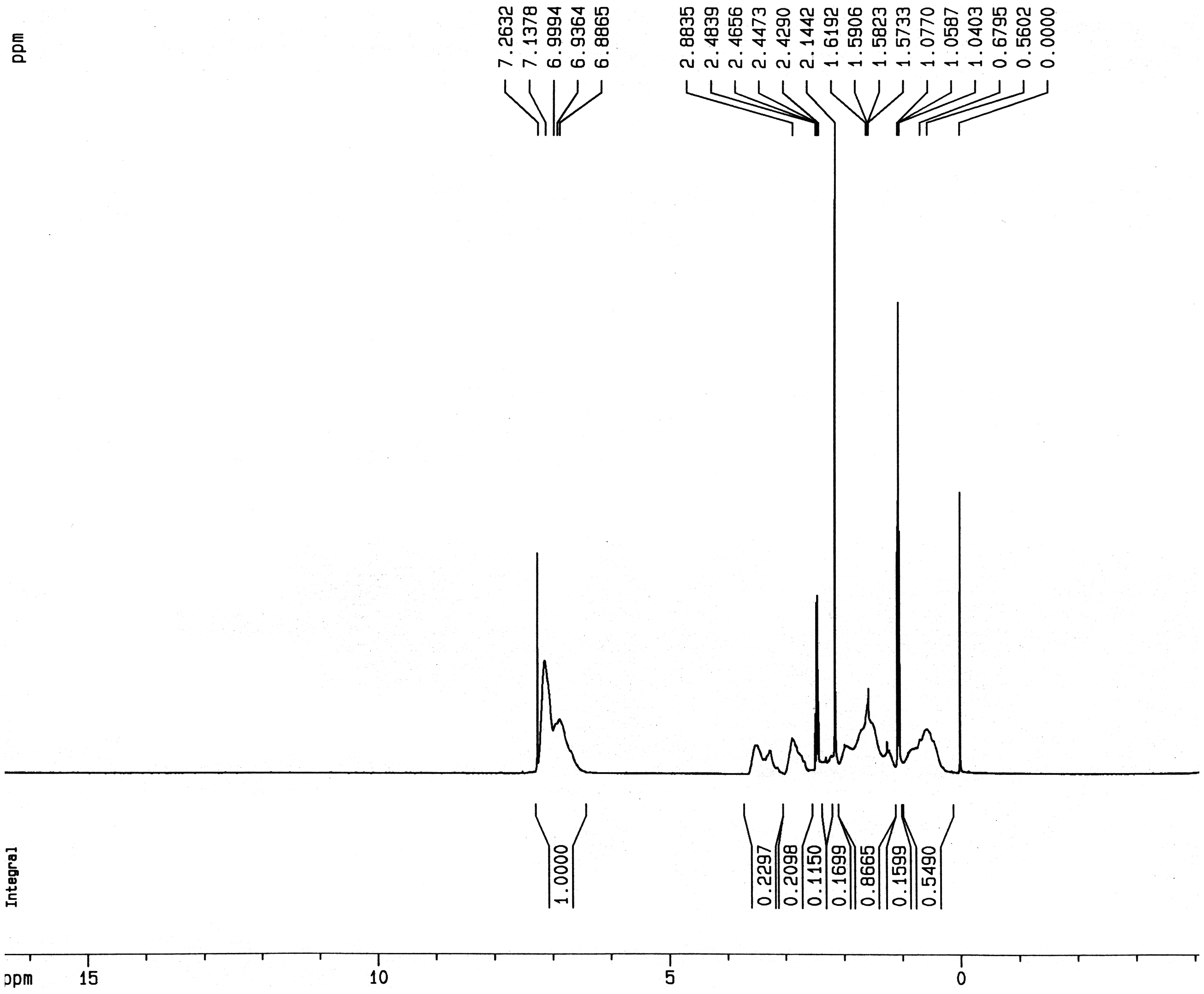
DSC thermogram of St/MMA beads showed two distinct peaks in which the first one (Tg = 95°C) is the glass transition temperature of copolymer, and the second one (nearly T = 117°C) is observed due to the long chain of MMA in the copolymer that is actually the glass transition temperature of PMMA (Figure 2). As it was concluded from H-NMR spectra, St–MMA copolymer is a random copolymer and it is possible that the chain length of one component in the copolymer structure is longer than the other one.
The DSC thermogram of St–MMA beads.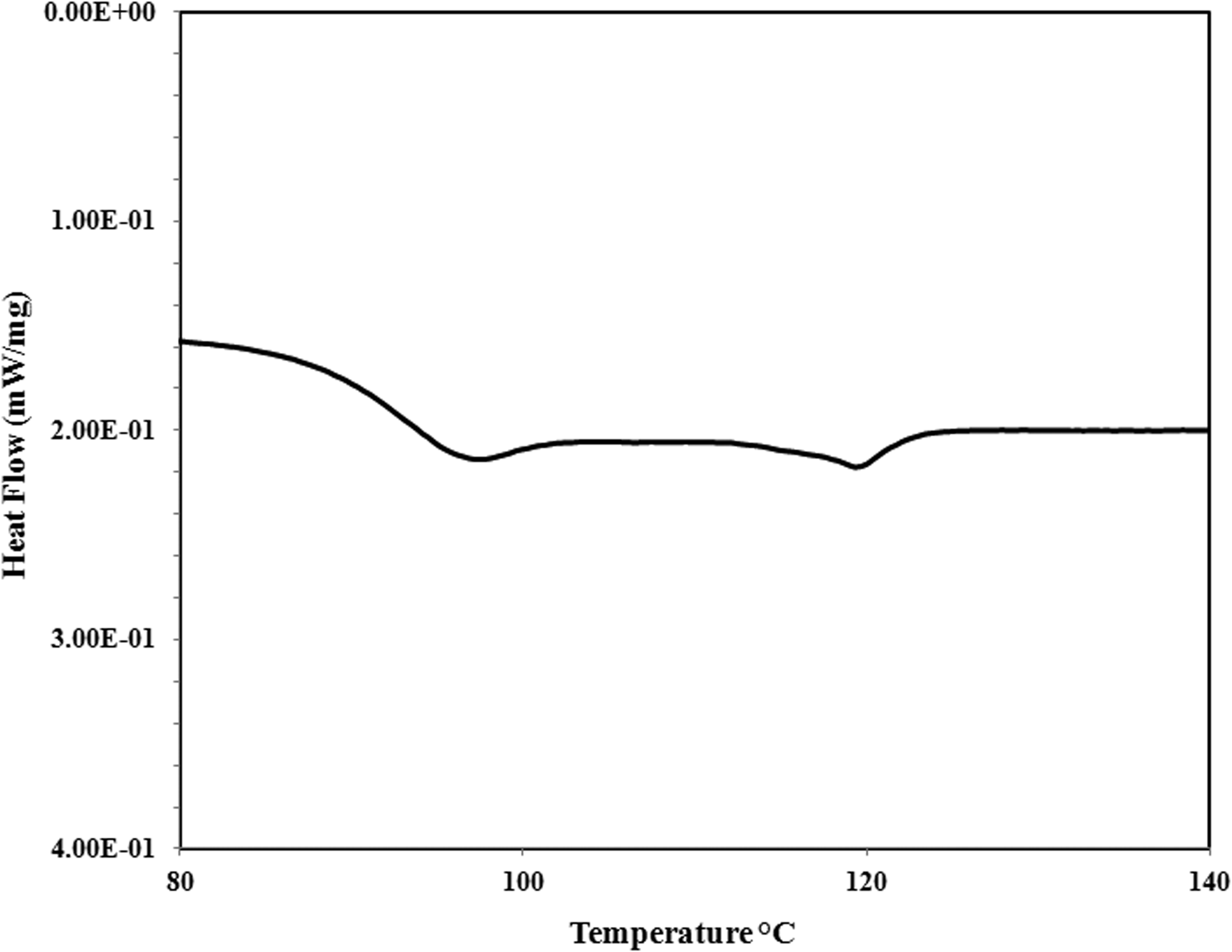
To determine MW and MWD from viscoelastic properties, the rheological experiments were conducted at T = 160°C, frequencies of 0.1–1000 per second and strain 1% under dry nitrogen purge. Thermal degradation was not suspected as measured by isothermal variation of G′ as a function of time. By performing strain sweeps on the disentangled and fully entangled samples, the linear viscoelastic region was obtained. Due to high sample stiffness and ease of loading of high MW samples, parallel plates were used instead of cone-and-plate geometry with a disk diameter of 25 mm and a sample thickness 1 mm. It has been experimentally verified that cone-and-plate and parallel plates gave identical results and η′ were calculated using standard equations incorporated in the rheometer software.
32
The data were analyzed with Rheometric Scientific software (Anton Paar). Disks were compression molded at 160°C and approximately 1000 psi. A vacuum port was fitted to the mold to prevent air dissolving in the disks while molding at high pressure; when the sample is melted in the rheometer at ambient pressure, the dissolved air would foam the sample. The viscoelastic characteristics of St–MMA copolymer is shown in Figure 3. The corresponding MW and MWD determined from the rheological experiments are as follows: MW = 278,000 (g/mol), Mn = 156,800 (g/mol) and PDI = 1.77.
Variation of complex viscosity, storage, and loss moduli vs frequency for St–MMA copolymer at T = 160°C.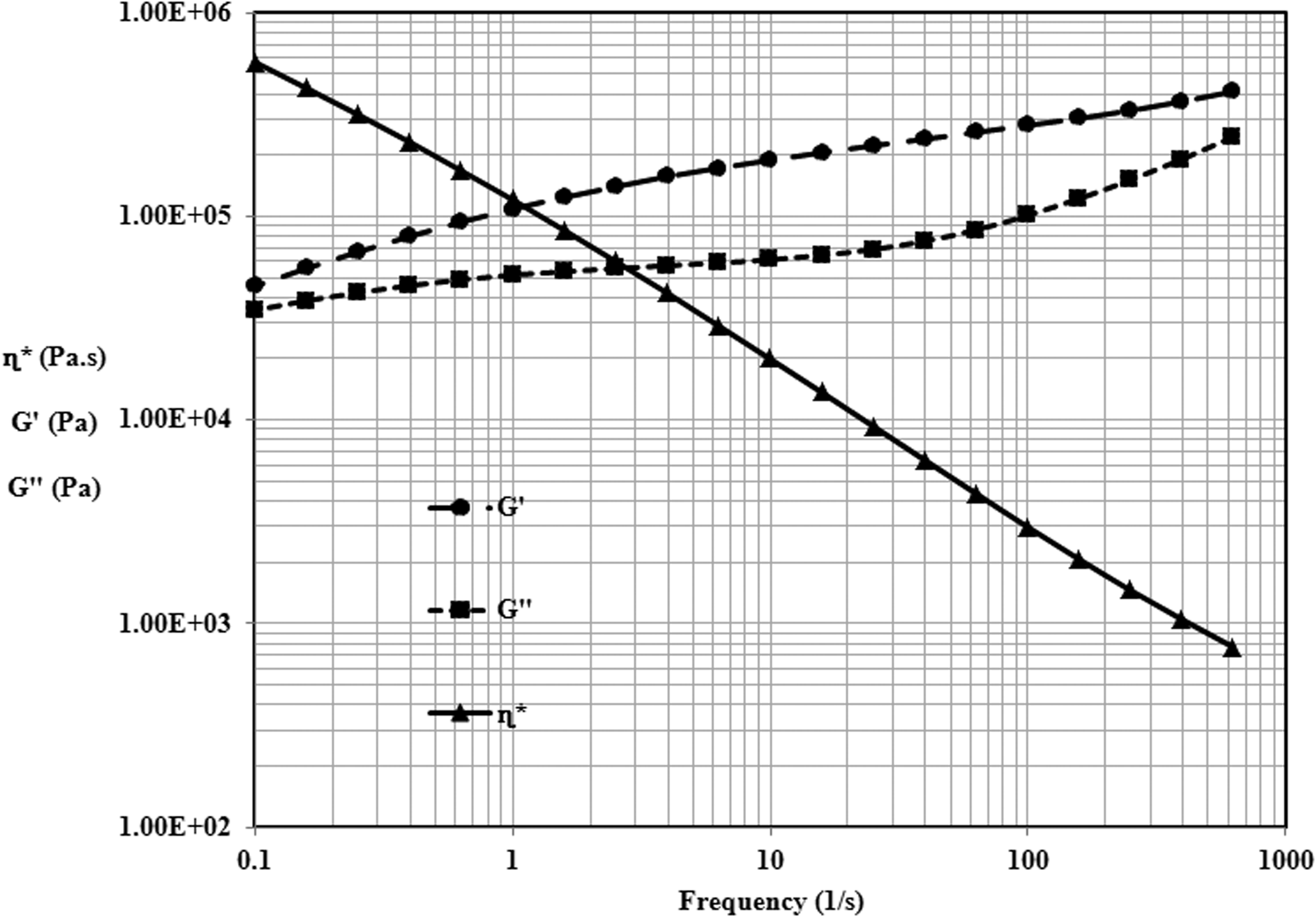
Foaming experiments
In this study, the effect of different expansion conditions, such as temperature, impregnation time, and impregnation pressure, on the foaming dynamics of St–MMA copolymer was investigated. In all experiments, the observation cell was charged with pentane at a determined time of pentane sorption into the copolymer particle with a size of 0.17–0.24 mm. It is clear that with increasing the particle size, the foaming conditions change. So, the same particle size (from the same mesh size of the sieved synthesized MMA–St copolymer particles) was used in all the experiments to eliminate the effect of particle size on the foaming behavior. After impregnation period and rapid pressure release, the particles are expanded in atmospheric pressure on the time scale of several minutes and their size reached a maximum size. In this study, the foaming and impregnation temperatures are the same. The foaming procedure of St–MMA copolymer particle with timpreg = 30 min, at 85°C and impregnation pressure 3 bar, is shown in Figure 4.
The foaming sequence of St–MMA copolymer particle after rapid pressure release at 85°C and ambient pressure (scale bar: 0.26 mm).
The expansion of particles was nearly complete after 130 s regardless of the impregnation time preceding the foaming. The effect of temperature and impregnation pressure on the foaming of St–MMA copolymer particles has been investigated at different temperatures 85°C, 95°C, and 105°C and distinct pressures 3 and 4 bar with timpreg = 30 min, as shown in Figure 5. The attempts to expand the impregnated particles below 80°C and pressure 3 bar, by rapid pressure release, were not successful.
The variation of foaming ratio of St–MMA copolymer particle vs time at different temperatures and pressures (The lines are used to guide eyes).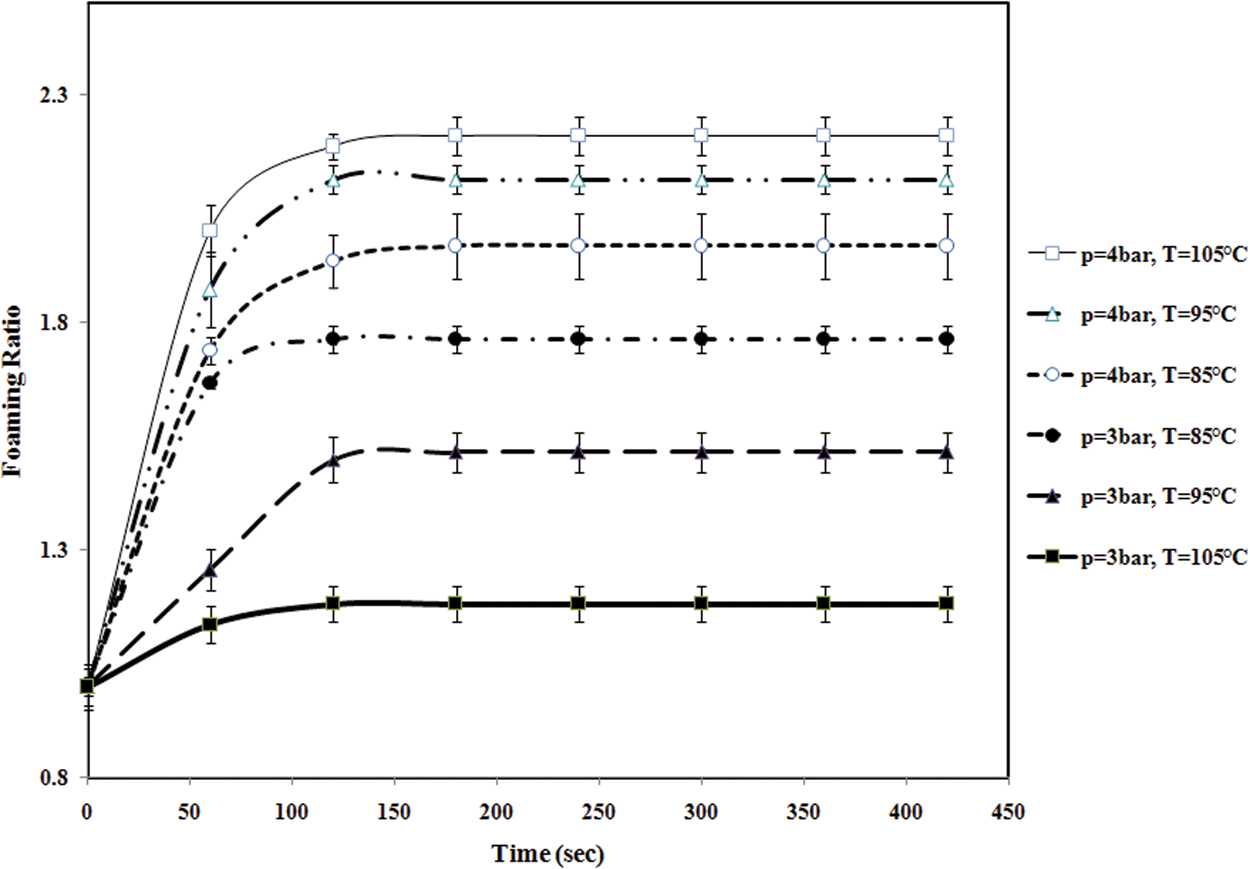
The variation of foaming ratio versus time at different temperatures and pressures is shown in Figure 5. As shown in this figure, at P = 3 bar, the foaming ratio decreases with increment in temperature.
Actually, when copolymer particles charge with pentane at different pressures, two distinct phenomena occurred. The first one is related to pentane solubility in St–MMA copolymer in which its solubility decreases with increment in temperature and the second one the diffusion of great amounts of pentane into the bulk of copolymer matrix. When the impregnation pressure is lower than 3 bar, decrease in pentane solubility with temperature mostly occurs and a significant fraction of pentane diffused out of the copolymer matrix and ultimately a small amount of the blowing agent contributed to the expansion process and diffusion into the foam cells. As well, this pressure (P = 3 bar) is not enough for pentane diffusion into the bulk of copolymer matrix; consequently, the expansion process at pressures lower than 3 bar is slightly affected by the second phenomenon. These conditions lead to the smaller expansion ratio. When pressure increases and reaches to greater than 4 bar, the expansion conditions change and the foaming ratio increases with temperature. In this stage, the foaming dynamics is governed by the second phenomenon. When temperature increases, the vapor pressure of occluded pentane in copolymer foam cells increases and the pressure difference between inside and outside of the cells increases and the driving force for cell expansion is exceeded. This behavior was confirmed with the SEM results at two temperatures (85°C and 105°C) and P = 4 bar, in Figure 6. The SEM images are shown from the cross-sectional area of the expanded particles. It can be concluded from these images in this copolymer composition that n-pentane cannot diffuse totally into the bulk of the copolymer and a core (solid) – shell (cellular) structure is formed.
The SEM images of the St–MMA copolymer at (a) T = 85°C and (b) T = 105°C, respectively, and P = 4 bar.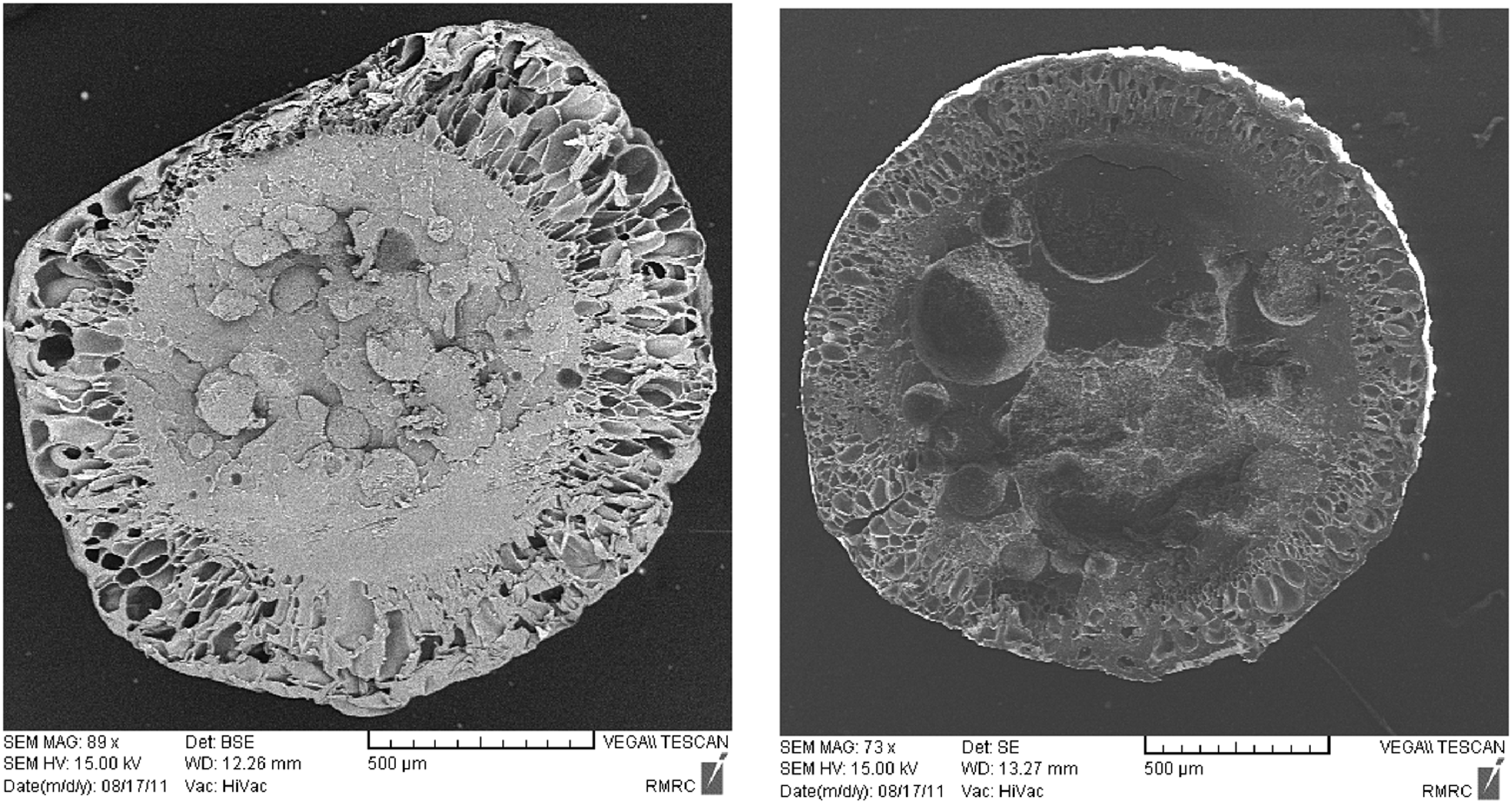
The maximum foaming ratio at different conditions is shown in Figure 7. As it is clear in these figures, at a pressure lower than 4 bar, the maximum foaming ratio decreases with increment in temperature but the contradictory behavior is observed at pressures higher than 4 bar.
Maximum expansion ratio of St–MMA copolymer at different temperatures and pressures.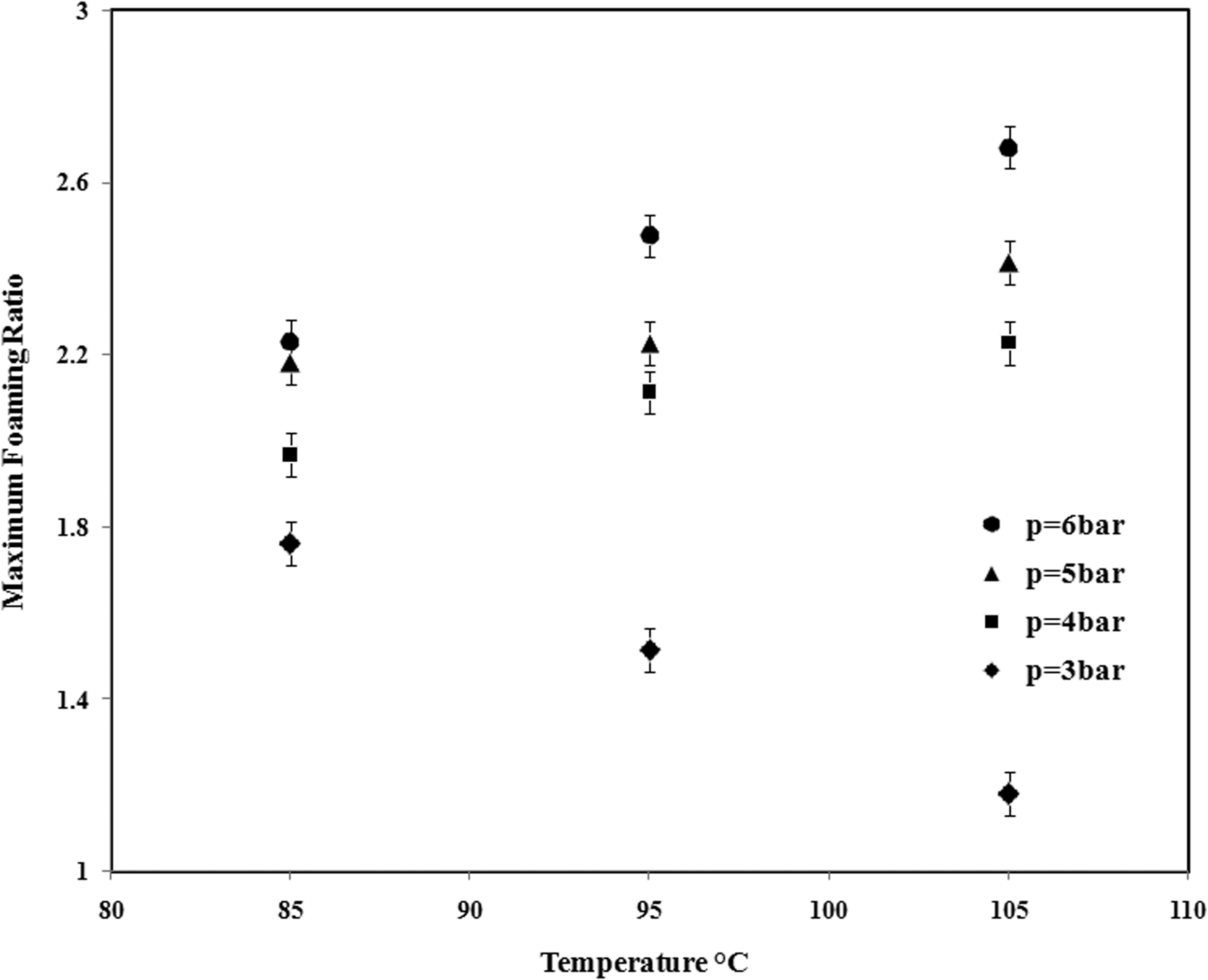
In Figure 8, the expansion rate at different pressures and T = 95°C are compared. As shown in this figure, with increase in the pressure, the expansion rate increases as well, because at the early stage of foaming, in high pressure, pentane diffusion into the cells is faster and copolymer particle expansion is more quick.
The rate of expansion of St–MMA copolymer at different pressures and T = 95°C.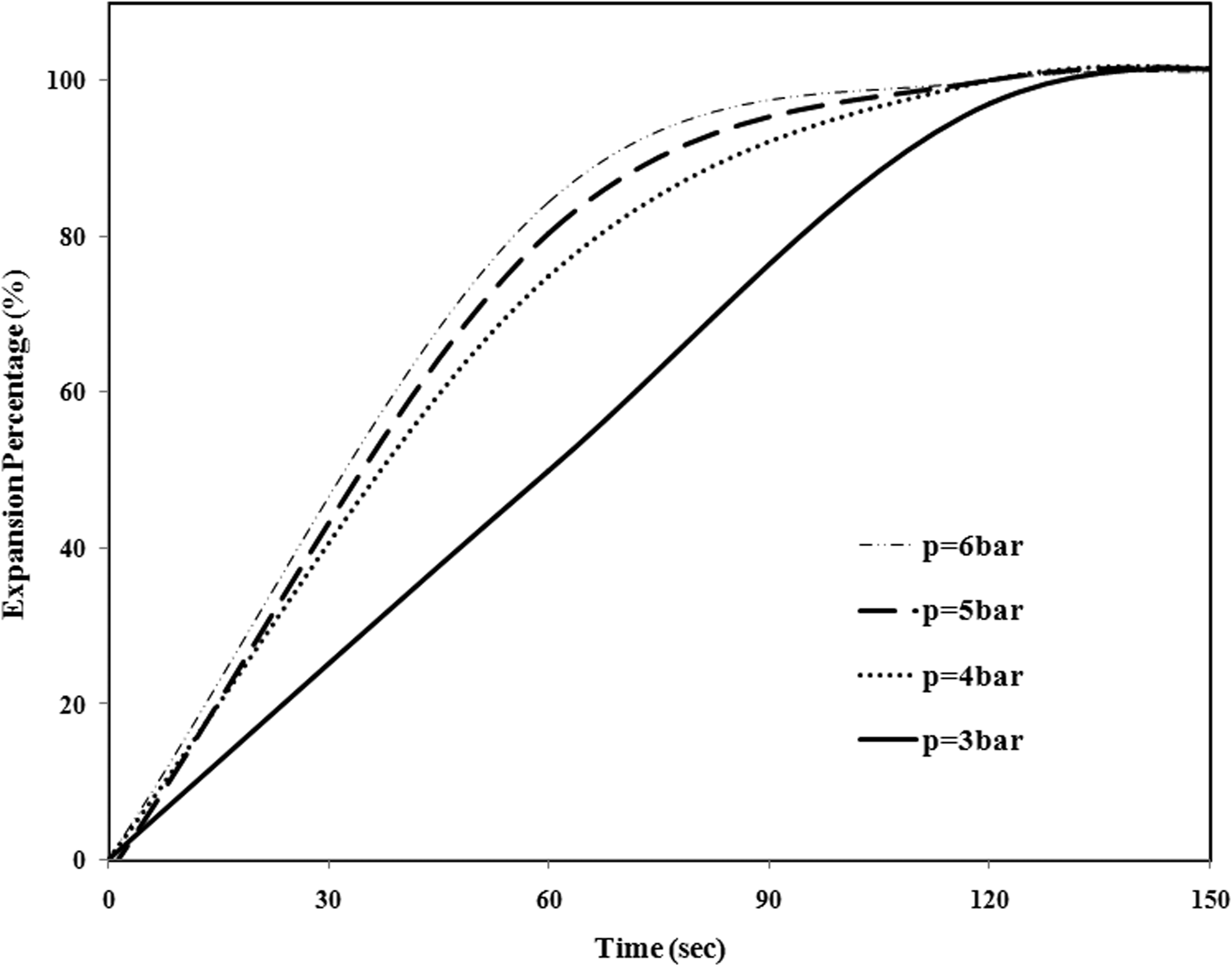
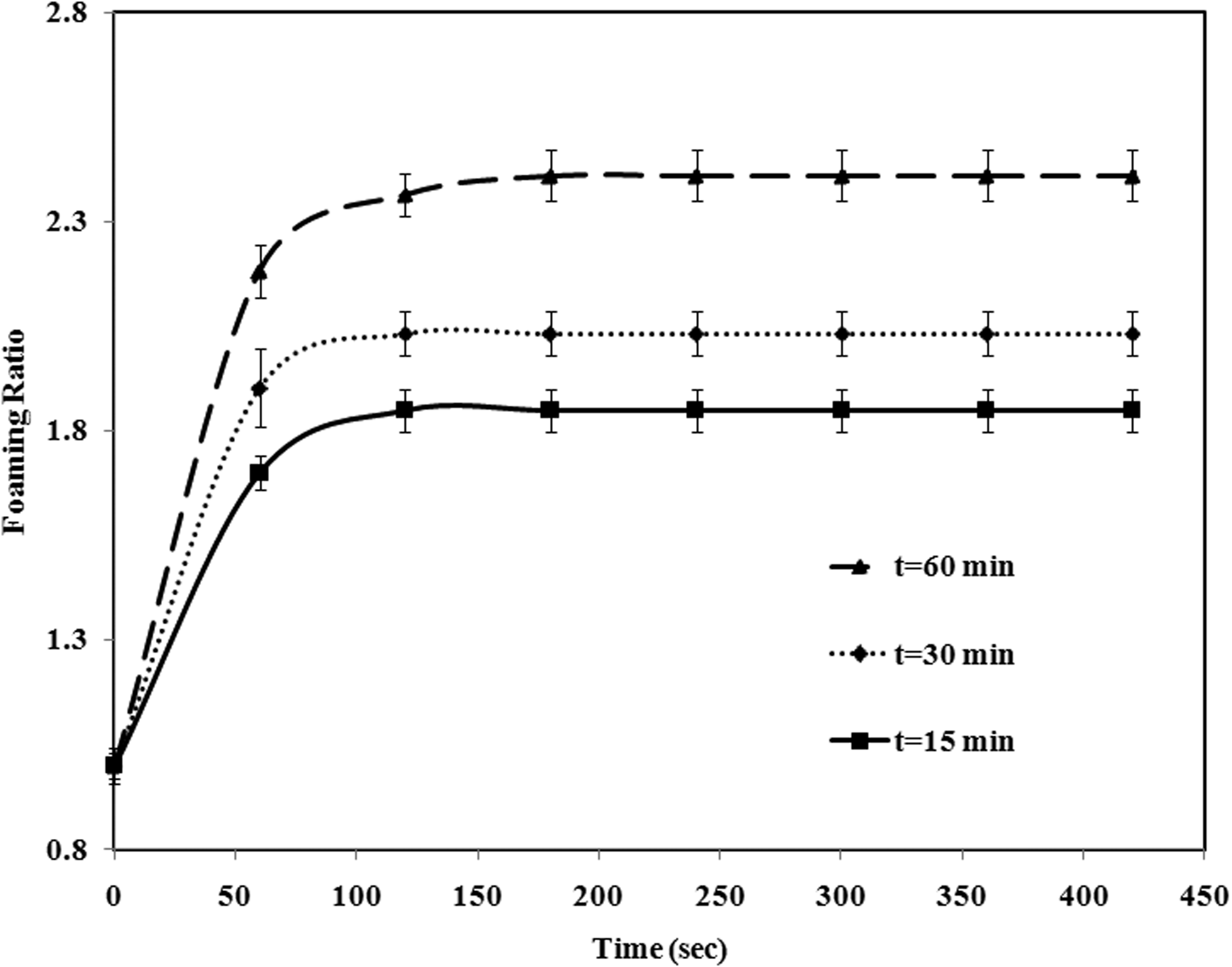
The effect of impregnation time on the expansion sequence and foaming ratio of St–MMA copolymer is shown in Figures 9 and 10. The particles were impregnated by pentane at an impregnation pressure of 4 bar and temperature 95°C for an impregnation period of timpreg = 15, 30, and 60 min. The foaming ratio (d/d0) increases with increase in the impregnation time, because at a higher impregnation time, pentane molecules have enough time to dissolve into the softened copolymer matrix as well as to diffuse into the foam cells in the copolymer particles. Therefore, at the early stage of foaming after rapid pressure release, the expansion process occurs due to the before-mentioned phenomena.
Foaming sequence of St–MMA copolymer at different impregnation times, P = 4 bar and T = 95°C. Notes: Columns a, b and c are impregnation times 15, 30, and 60 min, respectively. Scale bar: 0.26 mm. Effect of impregnation time on foaming ratio of St–MMA copolymer at T = 95°C and P = 4 bar.
Conclusion
In this study, the foaming dynamics of St–MMA copolymer were investigated via visual batch foaming apparatus. St–MMA copolymer particles were synthesized by suspension polymerization at an weight ratio of St/MMA 53/47. The synthesized copolymer particles impregnated by pentane and then foaming dynamics were recorded as the sequence of images of expanding particles located in the pressure cell. The effect of different foaming conditions, such as temperature, impregnation time, and impregnation pressure, on the expansion ratio of the foam was investigated. It was concluded that in the impregnation stage, two distinct phenomena are observed at different impregnation pressures. The first one, which occurred at pressures lower 4 bar, is related to pentane solubility in St–MMA copolymer which decreases with temperature increment and the second one takes place at pressures higher than 4 bar when large amounts of pentane diffuse into the copolymer bulk and fill the cells after rapid pressure release which leads to the higher foaming ratio. Thus, due to these contradictory mechanisms, at pressures lower than 4 bar, the growing trend was observed for foaming ratio with increasing impregnation pressure and time; however, it decreased with temperature rise, but the conflicting behavior was observed at pressures higher than 4 bar with temperature rising.