
Abstract
Poly (lactic acid) (PLA) foams prepared by supercritical CO2 foaming offer a sustainable route toward lightweight biodegradable materials, yet their cellular uniformity and mechanical stability are strongly limited by the poorly controlled coupling between crystallization and gas expansion. This study establishes a thermodynamic–kinetic framework to clarify how CO2-induced plasticization regulates crystallization kinetics, bubble nucleation, and structural fixation in PLA foams. DSC, SAXS, Avrami kinetic analysis, SEM morphology statistics, and compression testing reveal that dissolved CO2 lowers the crystallization activation energy from
Introduction
Biodegradable polymer foams have attracted extensive attention in packaging, cushioning, and biomedical applications due to their lightweight nature, 1 shock absorption capability, 2 and environmentally friendly degradation behavior. 3 Among them, poly (lactic acid) (PLA) stands out as one of the most commercially promising biodegradable polymers thanks to its good mechanical strength and renewable origin. 4 However, the formation of a uniform and stable cellular structure in PLA-based foams remains challenging. 5 PLA exhibits intrinsic brittleness and relatively slow crystallization kinetics, which hinder the effective stabilization of growing cells during foaming. 6 Supercritical CO2 (scCO2) foaming technology has provided an efficient solution with precise control over gas dissolution, nucleation, 7 and cell expansion processes while avoiding organic solvents. 8 Even so, optimizing cell morphology in biodegradable foams still requires deeper understanding of the molecular-level structural evolution during processing. 9
Current studies indicate that crystallization behavior plays a crucial role in the formation and stabilization of cell structures during supercritical foaming10. The crystalline phase affects gas diffusion, determines the availability of nucleation sites, 11 and provides mechanical support to freeze cells during expansion. 12 For PLA, the presence of scCO2 can modify its crystallization kinetics through plasticization and heterogeneous nucleation effects. 13 The degree of crystallinity, crystal growth rate, and spherulite morphology all influence the final foam structure, 14 including the average cell size, cell density, and cell wall thickness. 15 Yang et al. further demonstrated that both crystallinity and crystalline morphology significantly affect the cellular structure of PLA foams during scCO2 foaming, and that appropriate crystalline morphology can promote the formation of more uniform closed-cell structures. 16 Xiang et al. reported that microfibrillated and hierarchically organized PLA crystalline structures are effective in broadening the foaming window and improving mechanical robustness, indicating that crystal architecture can act as an active structural regulator rather than a passive solid phase. 17 These studies provide important evidence that PLA crystal structure has a direct influence on cell nucleation, growth restriction, and foam morphology stabilization. Nevertheless, most existing research has focused on final crystallinity or specific crystalline morphology, while the quantitative correlation between crystallization kinetic parameters and structural characteristics of PLA foams remains insufficiently clarified. 18 A systematic investigation is required to reveal how crystallization dynamics interacts with process variables such as saturation pressure, foaming temperature, and depressurization rate.19–27
This work established a clear structure–processing–crystallization relationship for PLA foams produced by supercritical CO2 foaming. By regulating PLA crystallization through thermal pretreatment and adjusting foaming conditions to alter CO2-induced crystallization kinetics, the evolution of crystalline morphology and its effects on cell nucleation and growth will be examined. Differential scanning calorimetry (DSC), scanning electron microscopy (SEM), and Avrami-based kinetic models will be employed to quantify crystallization characteristics and correlate them with changes in foam cellular structure. The central goal is to identify controllable crystallization kinetic parameters that enable fine-tuned cell morphology, providing a mechanistic foundation for designing high-performance biodegradable polymer foams with tailored functionality.
Experimental and methodology
Materials and characterization techniques
Poly (lactic acid) (PLA) was selected as the primary biodegradable polymer in this study due to its commercial accessibility and clear crystallization behavior. The material was processed into pelletized form and dried prior to foaming to minimize moisture-induced degradation. Melt flow characteristics were evaluated using a capillary rheometer to determine flow stability and viscosity under foaming-relevant thermal conditions, which are essential for maintaining structural integrity during cell growth. To investigate crystallization kinetics and structural evolution, a combination of thermal and structural characterization techniques was employed. Differential scanning calorimetry (DSC) was utilized to analyze non-isothermal and isothermal crystallization behavior, enabling calculation of crystallization parameters based on Avrami and Lauritzen–Hoffman models. Polarized optical microscopy (POM) provided insights into spherulite nucleation density and growth dynamics under controlled heating and cooling rates. Small- and wide-angle X-ray scattering (SAXS/WAXS) was applied in in-situ mode to monitor lamellar thickening, long period evolution, and crystal orientation during gas penetration and crystallization. Additionally, Fourier-transform infrared spectroscopy (FTIR) was used to examine potential CO2-induced chemical interactions, while gel permeation chromatography (GPC) confirmed molecular weight stability after processing. Density and porosity changes before and after foaming were recorded using the Archimedes method for quantitative correlation with cellular structure characteristics.
Supercritical CO2 foaming procedure
Supercritical CO2 foaming experiments were conducted in a high-pressure batch reactor with precise control over saturation pressure and temperature. Commercial PLA pellets were first dried in a vacuum oven at 60°C for 12 h to remove residual moisture. Dried PLA pellets (10.0 g for each batch) were compression-molded into rectangular specimens with dimensions of
For each foaming experiment, approximately 2.0 g of PLA specimens were placed in the reactor. Liquid CO2 was introduced using a high-pressure pump until the preset saturation pressure was reached. The samples were saturated at selected temperatures and pressures to obtain target CO2 uptake levels of 0 wt%, 5 wt%, and 10 wt%. After saturation equilibrium was reached, rapid depressurization was performed by opening the pressure-release valve within 3–5 s to induce cell nucleation and growth. The foamed samples were immediately cooled to room temperature to stabilize the cellular morphology. The residual CO2 was allowed to desorb naturally under ambient conditions for 24 h before structural characterization.
Accordingly, these three CO2 contents were designed to represent three characteristic states of PLA during foaming: non-plasticized PLA, moderately plasticized PLA, and highly plasticized PLA. This design enables systematic evaluation of how CO2 dissolution affects crystallization kinetics, gas-cell nucleation, and final foam morphology. CO2 content was determined using the mass difference method:

Rapid pressure release triggered phase separation, enabling bubble formation throughout the matrix, while subsequent cooling stabilized the cellular architecture by preventing post-growth coalescence and gas escape. During this process, crystallinity and crystallization kinetics were purposely regulated through thermal pretreatments and controlled cooling rates, creating different solid-phase constraints during foaming. Key variables included initial crystallinity, crystallization rate, foaming temperature relative to the glass transition region, and gas dissolution content, all of which influence nucleation density and cell expansion efficiency. After foaming, samples were stored under ambient conditions for structural relaxation prior to characterization.
Scanning electron microscopy (SEM) was used to evaluate cell morphology after cryogenic fracture, including average cell diameter, cell density, and wall thickness. Structural parameters were quantified through image analysis software to ensure statistical reliability. The combined experimental approach enabled the establishment of a direct relationship between crystallization dynamics and the formation of the final porous structure in PLA foams processed by supercritical CO2.
Microstructural characterization and crystallization kinetic modeling
The cellular morphology of the foamed PLA samples was examined through scanning electron microscopy (SEM). Samples were cryo-fractured in liquid nitrogen to preserve internal features and sputter-coated with a thin gold layer to improve conductivity. SEM images were analyzed using ImageJ software to quantify average cell diameter, size distribution width, and structural uniformity across different treatment conditions. The open-cell ratio was estimated by comparing fractured surface porosity with bulk density differences, enabling assessment of gas escape pathways during expansion. Foam density (ρf) was determined using the Archimedes displacement method. Based on measured density values, the cell number density (Nc) was calculated using the established relationship:

To evaluate crystallization behavior quantitatively, DSC data were fitted using the Avrami kinetic model, which describes the time-dependent variation of relative crystallinity:

The density of solid PLA
The sample density



Crystallization activation energy calculation
The apparent activation energy of non-isothermal crystallization was calculated from DSC cooling scans performed at different cooling rates. The DSC measurements were conducted using a TA Instruments Q2000 differential scanning calorimeter (TA Instruments, New Castle, DE, USA) under a nitrogen atmosphere with a flow rate of 50 mL min-1. Before testing, the instrument was calibrated using high-purity indium and zinc standards to ensure temperature and enthalpy accuracy. The temperature fluctuation during the test was controlled within ±0.1°C to minimize thermal measurement errors.
For each measurement, PLA samples of approximately 5–8 mg were sealed in aluminum pans. The samples were heated to 190°C at 10°C min-1 and held for 3 min to eliminate previous thermal history, followed by cooling to 30°C at controlled cooling rates of 5, 10, 15, and 20°C min-1. The crystallization peak temperature
The Kissinger method was used as the primary approach to determine the crystallization activation energy

To further validate the activation energy trend, the Flynn–Wall–Ozawa (FWO) method was also applied:

Data analysis strategy
The analytical strategy focused on establishing a mechanistic mapping between crystallization behavior and foam microstructure. Structural metrics such as cell density and wall thickness were evaluated in parallel with crystallization parameters, enabling visualization of how crystal growth influences nucleation density and cell development. Data were grouped based on crystallinity levels to identify threshold values beyond which crystal-induced structural constraints dominate bubble expansion. Emphasis was placed on the role of inter-spherulitic boundaries, where enhanced nucleation tends to occur due to local free-volume concentration, while high crystallinity regions impose a rigid confinement that restricts cell growth and increases structural uniformity.
Special attention was given to the competitive interactions between bubble nucleation sites and spherulite interfaces during the foaming stage. Regions experiencing rapid crystallization often exhibit delayed gas diffusion and reduced cell expansion due to reduced chain mobility. Conversely, moderate crystallization rates promote heterogeneous nucleation and improve the fineness of cell dispersion. By pairing SEM results with kinetic parameters extracted from Avrami analysis, a structure-to-process mapping was developed that distinguishes the relative contribution of nucleation acceleration and growth suppression. This integrative evaluation supports the identification of controllable crystallization regimes capable of tailoring foam morphology for targeted performance, such as enhanced mechanical stability or increased gas retention efficiency.
Results and discussion
Mechanistic interpretation of crystallization kinetics under CO2-Assisted conditions
The preliminary characterization results in Figure 1 provide the structural and thermal basis for analyzing crystallization-regulated PLA foaming behavior. The DSC thermogram in Figure 1(a) shows a typical thermal response of semi-crystalline PLA, with a glass transition at approximately 60.8°C, a distinct cold crystallization peak at 112.4°C, and a melting endotherm at 167.3°C. The presence of a pronounced cold crystallization peak indicates that the PLA matrix still contains a considerable amorphous fraction after thermal history treatment, suggesting that molecular chains retain sufficient mobility to rearrange during subsequent CO2-assisted foaming. The calculated crystallinity of 28.6% further confirms that the original PLA possesses an intermediate crystallization state, which is favorable for investigating the competition between crystal growth and gas-cell expansion. Preliminary characterization of PLA matrix and PLA foam. (a) DSC thermogram of PLA; (b) Polarized optical microscopy image of PLA spherulites; (c) SEM morphology of PLA foam; (d) Cell size distribution of PLA foam.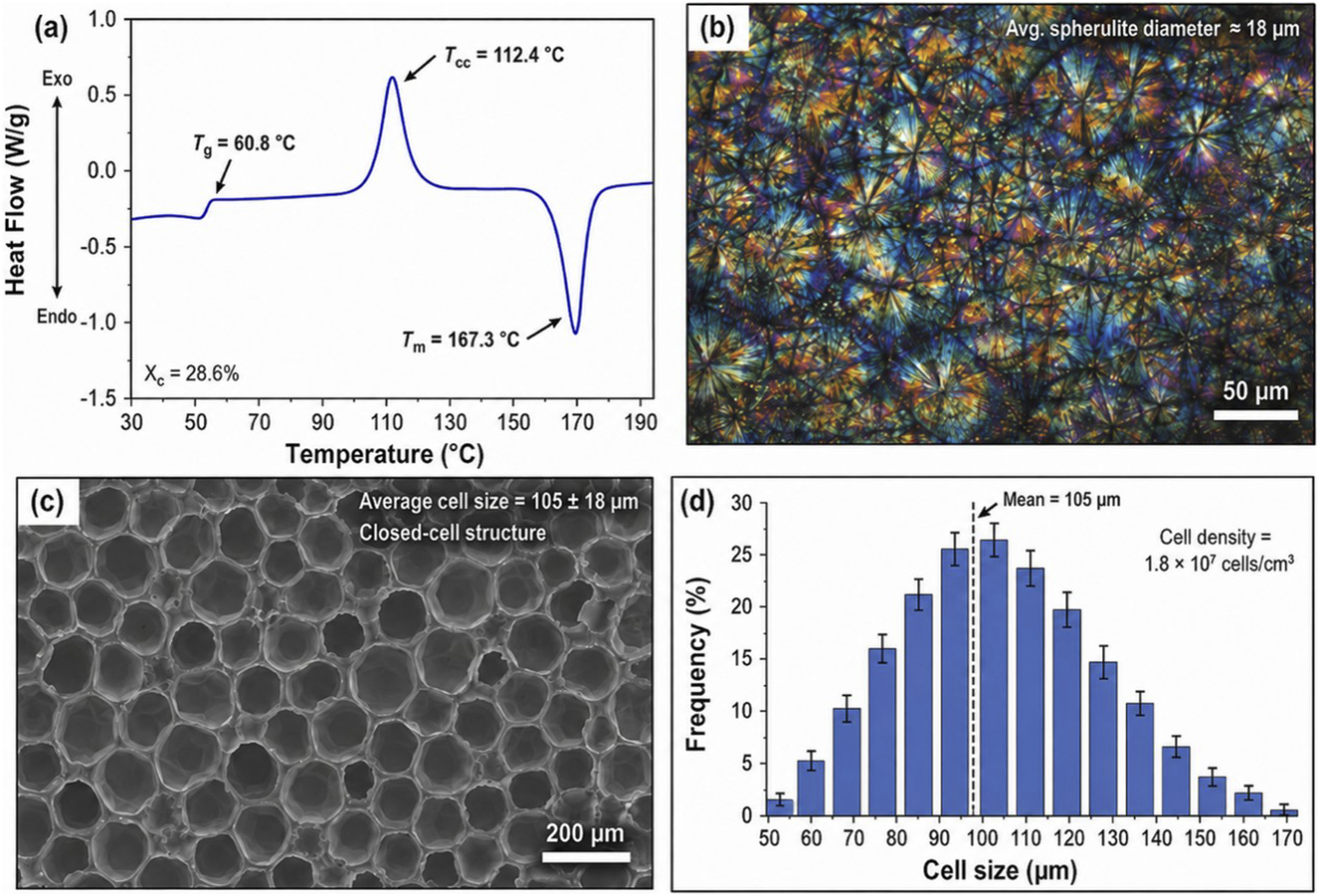
The polarized optical microscopy image in Figure 1(b) reveals densely packed spherulitic structures with clear birefringence, indicating that PLA develops well-defined crystalline domains under controlled crystallization conditions. The average spherulite diameter of approximately 18 μm suggests a relatively refined crystalline morphology rather than large isolated spherulites. Such a crystalline texture is important for foam formation because spherulite boundaries can act as heterogeneous nucleation sites during rapid depressurization. These interfacial regions may locally concentrate free volume and dissolved CO2, thereby lowering the energy barrier for bubble nucleation and contributing to a more uniform cellular structure.
The SEM image in Figure 1(c) shows that PLA foam prepared by supercritical CO2 foaming exhibits a predominantly closed-cell morphology. The cell walls are relatively continuous, and the pores are distributed without obvious large-scale collapse or severe coalescence. The measured average cell size of
The cell size distribution in Figure 1(d) further confirms the uniformity of the cellular architecture. Most pores are concentrated between approximately 80 and 130 μm, with a mean value of 105 μm and a calculated cell density of
The kinetic parameters extracted from Avrami fitting reveal that CO2 dissolution introduces a profound modulation of PLA crystallization pathways by simultaneously altering nucleation thermodynamics and chain transport kinetics. As presented in Figure 2(a), the crystallization rate constant ( Results of multi-method characterization of crystallization dynamics behavior; (a) crystallization rate constants under different CO2 contents; (b) variation of Avrami index with CO2 solubility; (c) non-isothermal crystallization activation energy; (d) relative crystallinity of DSC over time; (e) change of lamellar long-period structure of SAXS; (f) trend of crystal nucleus density with temperature increase.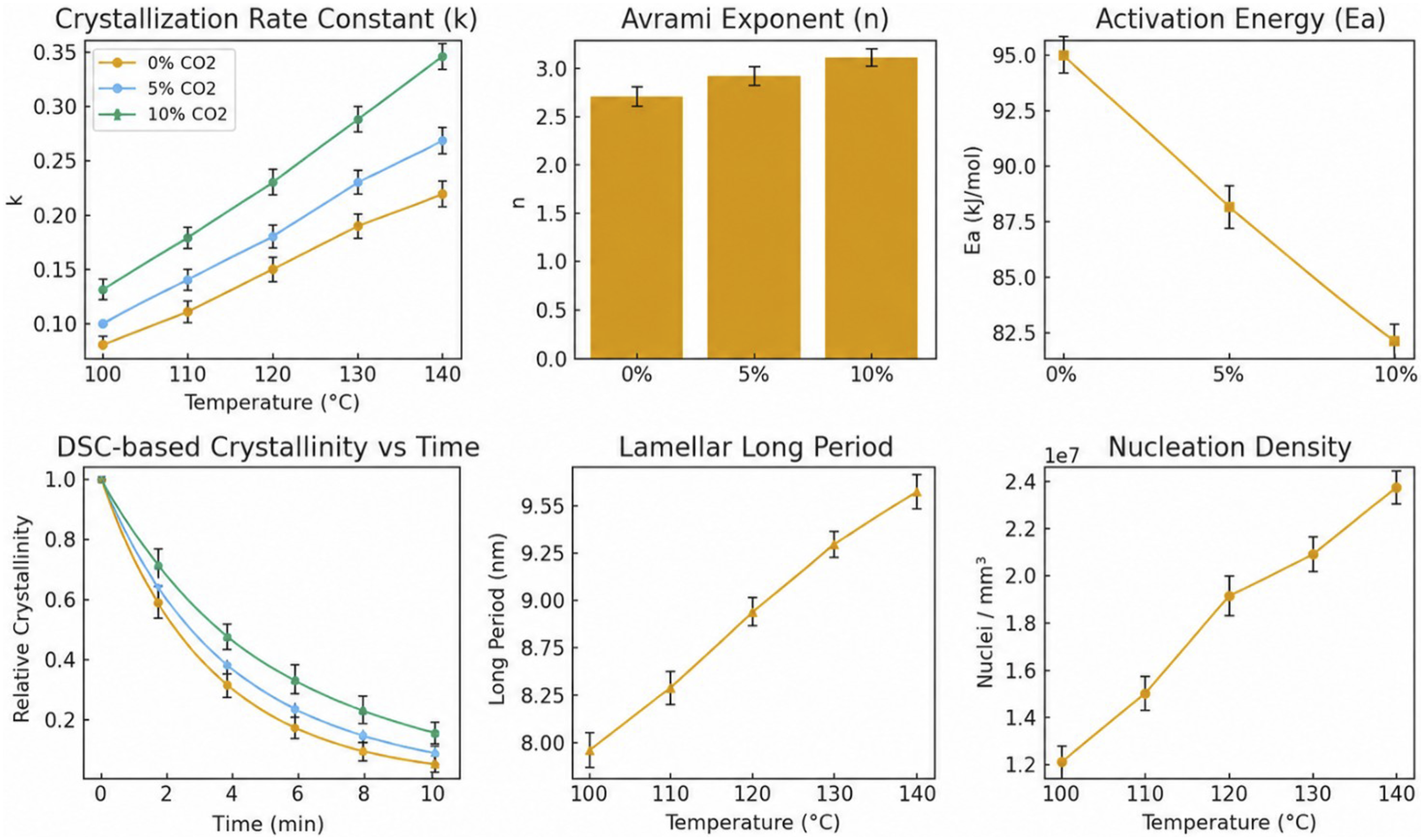
The variation in Avrami exponent provides additional evidence for the change in crystallization mechanism. As shown in Figure 2(b),
The reduction in non-isothermal crystallization activation energy from 95 to 82 kJ mol-1 (Figure 2(c)) further confirms the role of CO2 in lowering the energetic barrier for molecular ordering. This decrease indicates that PLA chains require less thermal energy to reorganize into crystalline structures when CO2 is dissolved in the matrix. SAXS results in Figure 2(e) show an increase in the lamellar long period with temperature, suggesting more developed lamellar structures under conditions where chain diffusion is sufficiently activated. These results collectively demonstrate that CO2 promotes crystallization not only by increasing molecular mobility but also by enabling more effective lamellar organization.
Integrating these structural and kinetic observations leads to a coherent mechanistic model. CO2-induced plasticization reduces the nucleation barrier and enhances chain diffusivity, while thermal energy promotes lamellar perfection, together yielding a dual-domain microstructure composed of highly nucleated yet mechanically coherent crystalline regions. This hybrid morphology offers both the nucleation density required for bubble initiation and the rigidity needed for structural stabilization during supercritical foaming. When the processing window is tuned such that crystallization proceeds within the optimal kinetic regime—sufficiently rapid to provide reinforcement but not so fast as to restrict gas expansion—the system achieves a self-regulated balance between crystallization and foaming dynamics. This thermodynamic–kinetic synchronization ultimately governs the formation of uniform cell architectures and underpins the superior mechanical and functional performance of CO2-foamed PLA.
The crystallization activation energy was calculated using the Kissinger method based on DSC cooling scans performed at different cooling rates. The fitting plots of
Evolution of foam cellular structure governed by crystallinity-induced nucleation dynamics
Quantitative analysis of SEM-derived structural parameters reveals a hierarchical co-regulation mechanism between crystalline phase development and cellular morphology evolution in CO2-foamed PLA. As shown in Figure 3(a) and (b), both nucleation density and final cell density increase nonlinearly with crystallinity, exhibiting a pronounced acceleration when crystallinity surpasses approximately 30%. This inflection reflects the onset of an interconnected spherulitic boundary network that functions as an energetically favorable heterogeneous nucleation scaffold during depressurization. The area fraction of such boundaries, increasing from 20% to 47% between 10% and 50% crystallinity (Figure 3(f)), provides dual functionality: it supplies additional interfacial sites for CO2-induced phase separation and simultaneously concentrates localized strain fields that catalyze bubble initiation. The formation of these interfacial microcavities effectively couples the solid-state crystallization front with gas-phase nucleation kinetics, leading to a more spatially homogeneous distribution of nuclei and consequently a finer, more isotropic pore morphology. Shows the variation law of cell structure with crystallinity; (a) Pore nucleation density; (b) Cell number density; (c) Average aperture; (d) Hole wall thickness; (e) Opening ratio; (f) Proportion of spherulite interface area.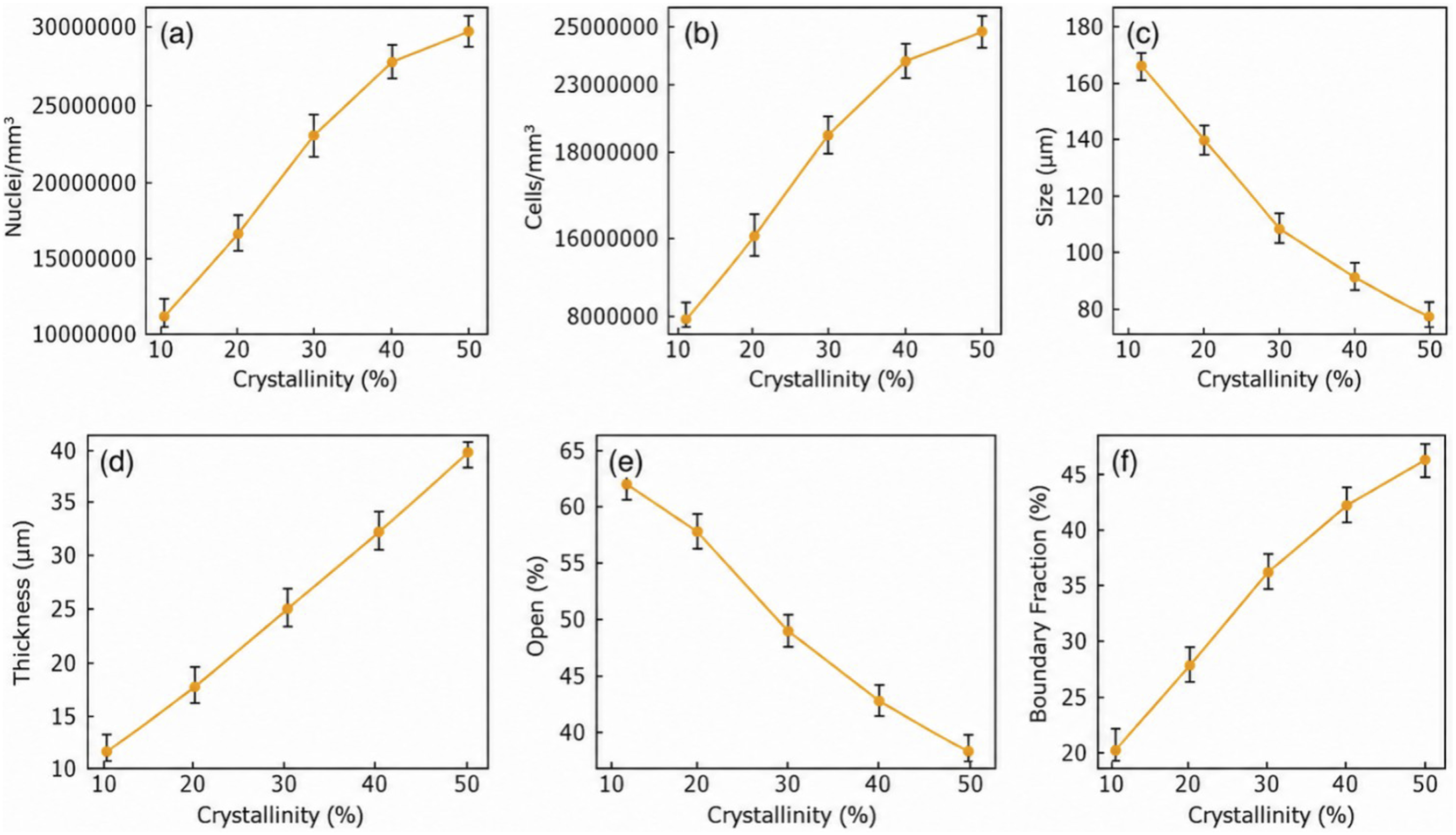
The decrease in average cell size shown in Figure 3(c) further confirms the role of crystallinity in limiting bubble growth. As crystallinity increases from 10% to 50%, the average cell size decreases from approximately 165–180 μm to below 80 μm, suggesting that bubble expansion becomes increasingly confined by the crystalline framework. At the same time, Figure 3(d) and (e) show an increase in cell wall thickness and a decrease in open-cell ratio, respectively. These trends indicate that crystalline lamellae reinforce the cell walls and suppress cell coalescence or rupture during expansion. Therefore, the crystalline phase contributes simultaneously to pore refinement and structural stabilization.
Integrating these observations suggests a dual-mechanism model in which crystallinity simultaneously governs nucleation thermodynamics and growth kinetics. Increased crystallinity elevates interfacial nucleation potential by expanding spherulitic surface area, while the resulting lamellar network imposes diffusion and mobility constraints that limit gas expansion. The interplay between these counteracting factors—nucleation enhancement versus growth inhibition—defines the morphological stability window of the foam. Optimal cellular architectures emerge within an intermediate crystallization regime, where nucleation density and matrix rigidity reach dynamic equilibrium. Within this range, bubble growth remains isotropic and well-distributed, yielding foams that combine high cell density, uniform pore size, and robust mechanical integrity. This crystallinity–structure coupling mechanism provides a unified framework for understanding how controlled crystallization kinetics dictates the multiscale organization and property evolution of biodegradable foams under supercritical conditions.
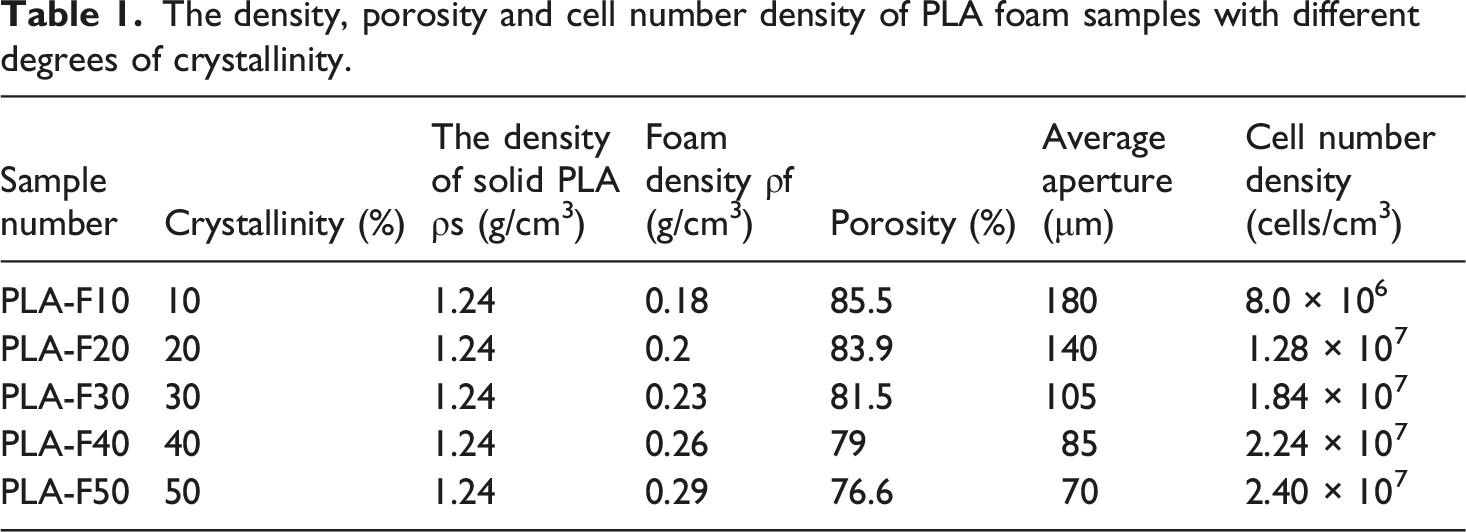
The supplemented density data provide a quantitative basis for evaluating the foam expansion and cellular structure. The density of solid PLA was measured as
The density, porosity and cell number density of PLA foam samples with different degrees of crystallinity.
Thermodynamic–kinetic coupling mechanism analysis
The progressive reduction in activation energy with increasing CO2 dissolution (Figure 4(a)) reflects a nontrivial thermodynamic softening of the PLA matrix, where CO2 molecules act as transient plasticizers that disrupt intermolecular entanglements and facilitate cooperative chain mobility. This melt-plasticization effect reduces the free-energy barrier for segmental rearrangement, enabling faster nucleation and lamellar ordering under lower thermal activation. Combined with the temperature-driven crystallinity enhancement (Figure 4(b)), the polymer evolves into a dual-phase dynamic state characterized by the coexistence of highly mobile amorphous chains and rapidly propagating crystalline fronts. Within this metastable region, crystallization no longer proceeds linearly with temperature but becomes self-accelerating due to the positive feedback between CO2-induced free-volume expansion and the release of latent crystallization heat. This thermodynamic–kinetic coupling effectively shifts the foaming regime from diffusion-limited to nucleation-controlled behavior during the early stages of bubble formation. The influence of the thermodynamics-dynamics coupling mechanism on the regulation of pore structure; (a) CO2-softening leads to a decrease in crystallization activation energy; (b) The relationship between temperature and crystallinity; (c) The average pore diameter decreases with the reduction of CO2 content; (d) The pore wall strength increases with the enhancement of crystallization; (e) The opening rate decreases with the increase of CO2; (f) The pore size uniformity improves; (g) DSC simulation shows that high CO2 accelerates crystallization; (h) SAXS layer-plate structure increases with temperature; (i) Three-dimensional schematic diagram shows the spatial constraining effect of the crystal interface on the bubbles.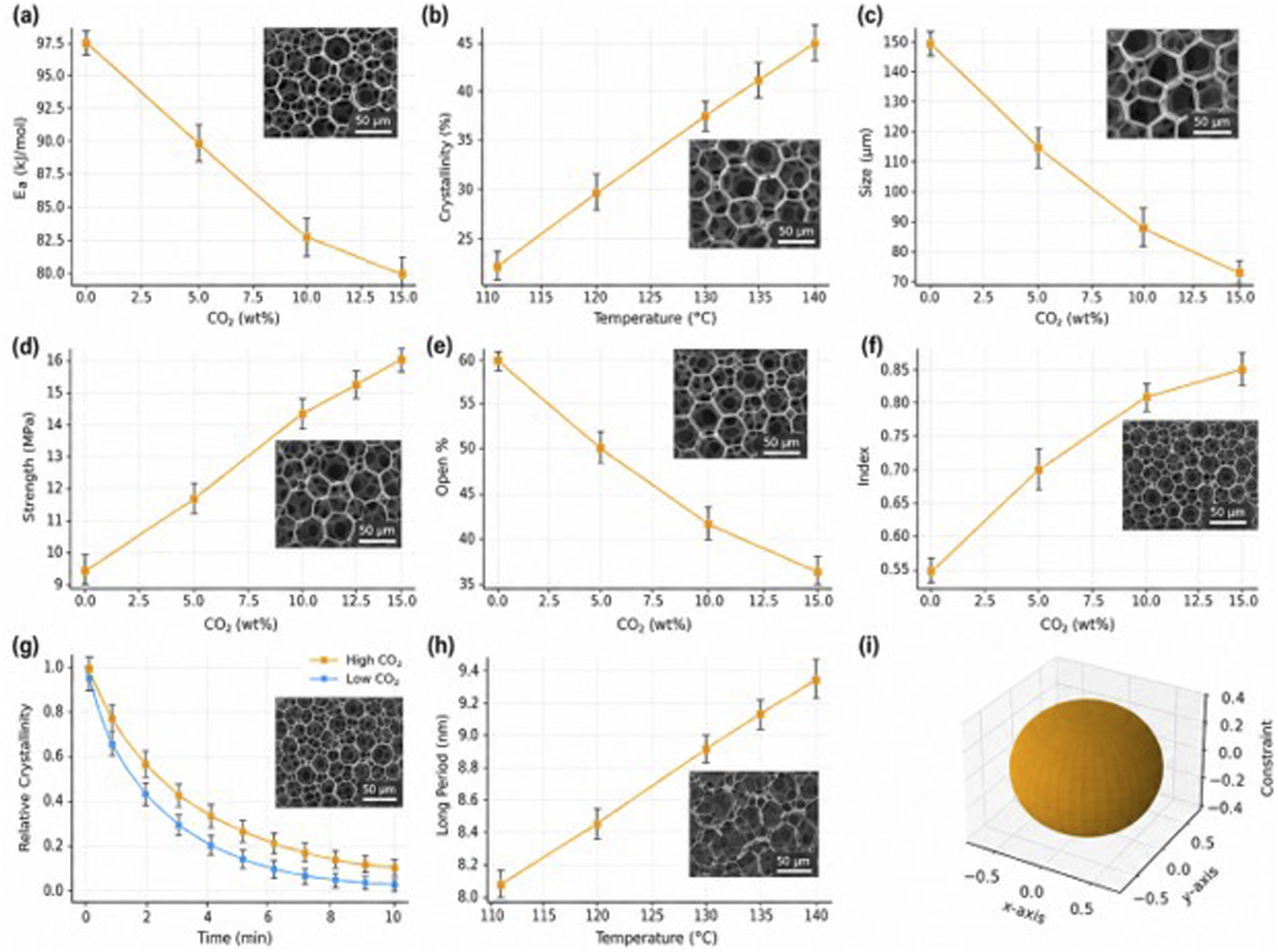
Microstructural evidence supports this coupling mechanism. At moderate CO2 uptake levels (5–10 wt%), the enhanced crystallization kinetics promote the emergence of smaller, more uniformly distributed cells (Figure 4(c) and (d)), which correlate with an increase in local nucleation density and suppression of uncontrolled coalescence. The concurrent reduction in open-cell ratio (Figure 3(e)) is attributed to the crystallization-induced mechanical reinforcement that stabilizes cell walls against tensile rupture during expansion. In this regime, the developing lamellar framework functions as an adaptive scaffold, dissipating internal gas stresses through elastic deformation and delaying the onset of wall collapse. The strengthening of the crystalline phase, evidenced by a significant rise in wall modulus (Figure 4(f)), signifies that the polymer transitions from a gas-dominated foam to a structurally percolated solid network, where crystallinity governs both load-bearing and diffusion resistance.
Once crystallization proceeds beyond the kinetic equilibrium threshold, the system behavior inverts. Excessively rapid crystal growth depletes the amorphous fraction, causing premature matrix solidification before gas pressure can equilibrate. The DSC profiles in Figure 4(g) exhibit shortened crystallization half-times at high CO2 content, confirming the kinetic overdrive of nucleation. Correspondingly, SAXS results (Figure 4(h)) reveal dense lamellar stacking and restricted long-period development, indicative of diffusion-limited crystal thickening. This over-crystallized regime induces morphological heterogeneity—uneven pore size distribution and localized collapse zones—arising from spatially asynchronous freezing of gas-filled domains. Such kinetic imbalance between solidification and expansion highlights the narrow processing tolerance inherent to CO2-foamed biodegradable systems.
The 3D schematic in Figure 4(i) conceptually illustrates this coupling behavior: advancing crystalline interfaces form a confining network that entraps bubbles within spherulitic “cages,” where interfacial stress gradients dictate the directionality of further growth. The optimal foaming window therefore exists at the intersection of two counteracting phenomena—CO2-induced plasticization that enhances crystallization kinetics and lamellar solidification that constrains expansion. Within this equilibrium zone, the structural evolution achieves synchronized crystallization and gas diffusion, yielding a foam microarchitecture that combines isotropic pore dispersion with high mechanical integrity. This thermodynamic–kinetic synergy establishes a predictive framework for designing next-generation biodegradable foams where microstructure and performance emerge from deliberately tuned crystallization dynamics.
Multiscale structure–performance relationships
The evolution of mechanical properties in PLA foams reflects a tightly interwoven relationship between multiscale structural organization and macroscopic deformation behavior. As crystallinity rises from 10% to 50%, a systematic increase in density is observed (Figure 5(a)), corresponding to the progressive transformation of the cellular architecture from a gas-dominated to a crystal-reinforced framework. This densification process signifies not merely a reduction of void fraction but a transition toward a more mechanically percolated morphology in which crystalline lamellae act as rigid junctions within the foam network. The associated refinement in average cell size and increase in lamellar connectivity enhance the local stiffness of the cell walls and improve stress transfer efficiency across adjacent cells. Consequently, the compressive modulus nearly triples within the same crystallinity range (Figure 5(b)), marking the transition from localized bending-dominated deformation to a more collective load-bearing response controlled by the crystalline skeleton. Characterization Results of the coupling relationship between multi-scale structure and mechanical properties; (a) Density varies with crystallinity; (b) Correlation between compressive modulus and density; (c) Energy absorption capacity increases with the increase of crystallinity; (d) Statistics of cell size distribution; (e) Compressive stress-strain response; (f) Negative correlation between aperture and modulus; (g) A three-dimensional schematic diagram shows the mechanical support mechanism of the crystalline network enhancing the cell structure.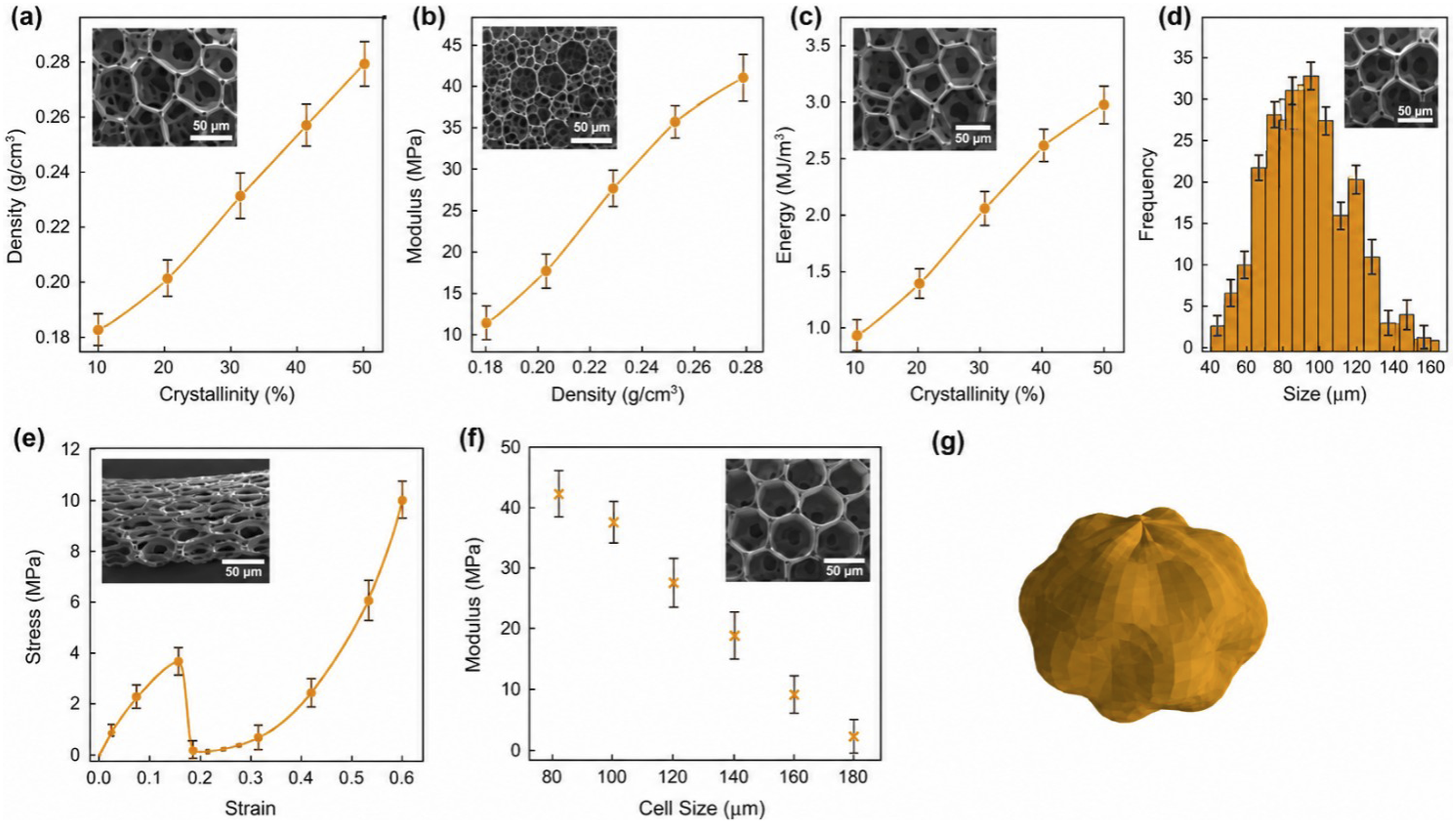
The strengthening mechanism is accompanied by a pronounced enhancement in energy dissipation capacity. As shown in Figure 5(c), the absorbed energy increases from 0.9 MJ m-3 to 3.0 MJ m-3 as crystallinity increases, indicating that the crystalline framework not only stiffens the structure but also delays the onset of catastrophic buckling. The stress–strain profiles (Figure 5(e)) reveal a qualitative change in deformation mode: amorphous foams display early collapse and brittle densification, whereas semi-crystalline foams sustain a stable plateau regime over an extended strain range. This mechanical resilience arises from the hierarchical microstructure, where lamellar reinforcement distributes local stress and prevents the coalescence of microcracks. Crystalline domains act as internal “anchors” that store and dissipate energy through elastic–plastic deformation of the surrounding amorphous matrix, transforming the collapse mechanism from abrupt failure to controlled, progressive compression. Morphological uniformity plays a crucial role in sustaining this mechanical balance. The pore-size distribution narrows markedly with increasing crystallinity (Figure 4(d)), reducing local stress gradients and enabling more homogeneous deformation. The negative correlation between average cell size and compressive modulus (Figure 5(f)) confirms that microstructural refinement drives macro-scale strengthening. Smaller, more uniform cells restrict strain localization, resulting in an energetically favorable load distribution that transforms individual cell-wall bending into coordinated network deformation. The 3D schematic (Figure 5(g)) illustrates this multiscale strengthening paradigm: crystalline lamellae percolate through the foam, forming a quasi-continuous load-bearing skeleton interlinked by gas-filled cells, effectively bridging nanoscale crystallinity with macroscale mechanical performance.
Integrating these results establishes a design framework linking molecular crystallization dynamics to bulk energy absorption performance. The optimal structural window is identified within the intermediate crystallinity regime (30–45%), where the competing effects of stiffness enhancement and ductility retention are balanced. In this range, the foam density (0.23–0.26 g cm-3) and cell size (<100 µm) collectively yield superior mechanical stability and energy dissipation efficiency. This coupling of microstructural regulation and macroscopic functionality underscores a generalizable strategy for tailoring biodegradable foams toward high-performance applications, including lightweight structural cores, shock-mitigating packaging, and bioresorbable scaffolds, where simultaneous stiffness, toughness, and uniformity are required.
Conclusions
This study establishes a quantitative crystallization–foaming relationship for PLA foams prepared by supercritical CO2 processing and demonstrates that crystallization kinetics plays a decisive role in regulating pore formation, cell-wall stabilization, and mechanical response. CO2-induced plasticization reduced the crystallization activation energy from
The optimized PLA foams exhibited clear mechanical advantages, including a compressive modulus of 28–36 MPa and an energy absorption capacity of 2.1–2.6 MJ m-3, confirming that controlled crystallization can simultaneously improve stiffness and impact-energy dissipation. These properties make the resulting foams suitable for biodegradable green packaging, lightweight cushioning materials, and shock-absorbing components where low density, uniform pore structure, and compression resistance are required. In addition, the ability to regulate pore size and cell-wall integrity through crystallization control may provide useful guidance for designing PLA-based biomedical scaffolds, especially in applications requiring interconnected porous architecture and adequate mechanical support. Overall, this work provides a practical processing strategy for producing structurally stable and mechanically reliable biodegradable polymer foams through the synchronized control of CO2 plasticization, crystallization kinetics, and cellular growth.
Conclusions
This study clarifies the role of crystallization kinetics in controlling cellular morphology and mechanical performance of PLA foams prepared by supercritical CO2 foaming. CO2-induced plasticization reduced the crystallization activation energy from
The obtained performance advantages indicate clear application potential in biodegradable green packaging, cushioning and shock-absorbing components, and PLA-based biomedical scaffolds. For packaging and cushioning, the low foam density and improved energy absorption are beneficial for lightweight protection. For biomedical scaffold design, the refined pore morphology and reinforced cell walls provide a basis for balancing porous architecture and mechanical support. This work therefore offers a specific and reproducible strategy for designing PLA foams with controlled cellular structure and application-oriented mechanical performance.
Footnotes
Acknowledgments
The authors thank all members of the research group for their support and helpful discussions.
Author contributions
Conceptualization, He Huang; Investigation, He Huang; Methodology, He Huang; Data curation, He Huang; Writing original draft, He Huang and Baohua Li; Writing review & editing, He Huang; Funding acquisition, He Huang; Project administration, He Huang and Liqin He; Supervision, Yue Chen. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.