
Abstract
Polyimide nanocomposites containing three different types of nanofillers were compared in terms of their thermal properties, morphology, and oxygen permeability. Organically modified hectorite clay (STN), functionalized hexadecylamine-graphene sheets, and an STN/hexadecylamine-graphene sheets complex were utilized as nanofillers in the fabrication of polyimide nanocomposite films. Hybrid films were obtained from blended solutions of the precursor polymer and the three types of nanofillers, and the filler content with respect to the polyimide was varied from 0 to 20 wt%. The variations in the properties of the polyimide matrix with respect to the filler content are discussed. Transmission electron microscopy photographs confirmed that most of the STN and hexadecylamine-graphene sheets were dispersed homogeneously throughout the matrix polymer on the nanoscale, although some agglomerate STN/hexadecylamine-graphene sheet complex particles were also formed. The coefficients of thermal expansion and the O2 transmission rates of the hybrid materials were found to improve linearly as the filler content of the polyimide matrix increased.
Introduction
Nanocomposites are a class of composite materials that contain ultrafine nanoscale fillers homogeneously dispersed throughout a polymer matrix. The nanoscale of the filler materials implies that nanocomposites possess a variety of properties that are superior to those of conventional composite materials since the interfacial adhesion is maximized. 1 Nanoscale composites containing polymers paired with clays or graphene-based materials have recently been the focus of extensive study.2–6
Clays possess sandwich-type structures that typically consist of one octahedral Al sheet and multiple tetrahedral Si sheets—clays possessing this typical structure are called phyllosilicates.7,8 Many forms of phyllosilicates exist, such as kaolinite, montmorillonite, hectorite, saponite, and synthetic mica. In general, hydrophilic clays are not compatible with most polymers and therefore require organic treatments in order to become organophilic. A common method of organic treatment is the ion exchange of the cations within the clay with organic ammonium cations. 9 Organically modified hectorite (STN) was chosen for our investigation into the synthesis of clay/polymer nanocomposites. STN consists of stacked silicate sheets with lengths of approximately 46 nm and thicknesses of 1 nm. STN has been widely used as a reinforcing filler in polymeric matrices as a result of its excellent mechanical, electrical, and thermal properties. 10
Graphene tends to aggregate or restack as a result of its strong stacking tendency and high cohesive energy. Additionally, the hydrophobic nature and high specific surface area of graphene renders it insoluble in a variety of organic solvents. Therefore, a key challenge in the preparation and processing of graphene-based composites is the prevention of aggregation. Functionalization of the graphene surface introduces reactive moieties that disrupt the bundle structure, and could potentially allow one to obtain individual sheets.11–13 This functionalization involves the attachment of functional moieties to the open ends and walls of graphene in order to improve the solubility and dispersibility of the graphene sheets (GSs). 14 As a result, one of the best methods for achieving homogeneous graphene dispersion throughout a polymer matrix is by using functionalized graphene sheets (FGSs). 15 This process involves functionalizing the graphene with polymers that are structurally similar to the matrix polymer in order to ensure that the dispersed graphene is compatible with the polymer matrix, thus preventing any microscopic phase separation in the nanocomposites. These FGSs also exhibit improved dispersion in solvents and polymers. Furthermore, covalent functionalization may provide the means for engineering a GS/polymer interface and subsequently optimizing the properties of the composite material. 16
Although traditional composite structures typically contain a significant amount (∼40 wt%) of filler bound within a polymer matrix, dramatic changes in the properties of the material are also possible at low loading amounts of nanofillers (<20 wt%) such as exfoliated nanoclays, carbon nanotubes, and GSs in the nanocomposites.17–19 This performance is achieved not only by utilizing the inherent properties of the nanofiller but also by optimizing the dispersion, interface chemistry, and nanoscale morphology in order to take advantage of the enormous surface area per unit volume that the nanofillers possess (the theoretical limits are 760 m2/g for clay and 2630 m2/g for graphene).20,21 These extra properties indicate that clay and graphene complex nanofillers would likely have significant potential for improving the thermal properties and gas barrier properties of various polymers through synergistic effects. 22
The thermal stability of nanofiller components such as clay and graphene is of major importance because many polymer composites are either melt-blended or intercalated at high temperatures in order to yield the corresponding nanosized composites. 23 The introduction of filler components into organic polymers can improve their thermal degradation stabilities. Nanofillers add thermal stability to the composites owing to the thermal isolation effect of inorganic sheets and the mass transport barrier they provide to the volatile products generated during thermal decomposition.24,25
The mobility of the polymer chain segments of polymer nanocomposites is clearly different from that of pure polymers because of the confined geometry, which affects the gas permeability. Two main factors are responsible for permeability reduction26–28: (i) the polymer chain-segment immobility and (ii) the detour ratio, which is defined as the ratio of the film thickness in the nominal diffusion flow direction to the average length of the tortuous diffusion distance between the nanofiller layers.
Aromatic polyimides (PIs) have been evaluated for use in microelectronics, aerospace, and military applications.29,30 Hence, PIs are high-performance polymeric materials characterized by their outstanding properties. PIs are also considered to be among the most important super-engineering materials because of their superior mechanical properties at elevated temperatures owing to their thermal stability. 31 Recently, much research effort has been devoted to developing high-performance PI materials with excellent thermal properties and gas permeation. 32
The aim of this investigation was to obtain experimental characterization data of the intermolecular interactions between the organic components in the nanofillers and the PI matrix polymer. Another objective was to evaluate the effects of varying the filler content of the PI hybrid films on the composite properties.
This manuscript discusses the thermal properties, morphology, and oxygen permeation of PI nanocomposites containing three different types of nanofillers: organically modified STN, functionalized hexadecylamine-GSs (HDA-GSs), and an STN/HDA-GS complex. STN/HDA-GS complex nanofillers containing two different layers are interstratified stacked together and exhibit interesting properties. Usually the properties of the composites are not the algebraic average of the properties of the individual layered nanofillers.
In the present study, we prepared hybrid films containing PI and an appropriate amount of filler (≤20 wt%) and examined their properties as a function of the filler content. PI is based on the reaction of 4,4′-biphthalic anhydride (BPA) and bis(4-aminophenyl) sulfide (APS). The thermal and oxygen barrier properties of the hybrids were also studied, in addition to the form of the film as a function of the filler content in the PI matrix polymer. We also investigated the effects of filler loading on the morphology of the PI hybrid films.
Experimental
Materials
All reagents were purchased from TCI (Tokyo, Japan) and Aldrich Chemical Co. (Seoul, Korea). BPA and APS were obtained from TCI and used as received. N,N′-Dimethylacetamide (DMAc) was purified and dried over molecular sieves prior to use. All other reagents were used without further purification. Natural flake graphite with a 75-mesh particle size was purchased from Aldrich Chemical Co. (Yongin, Korea), and STN clay was supplied by CO-OP Ltd. (Tokyo, Japan). The cation exchange capacity of STN was found to be in the range of 78 meq/100 g. The chemical structure of STN is shown in Scheme 1.
Chemical structure of STN.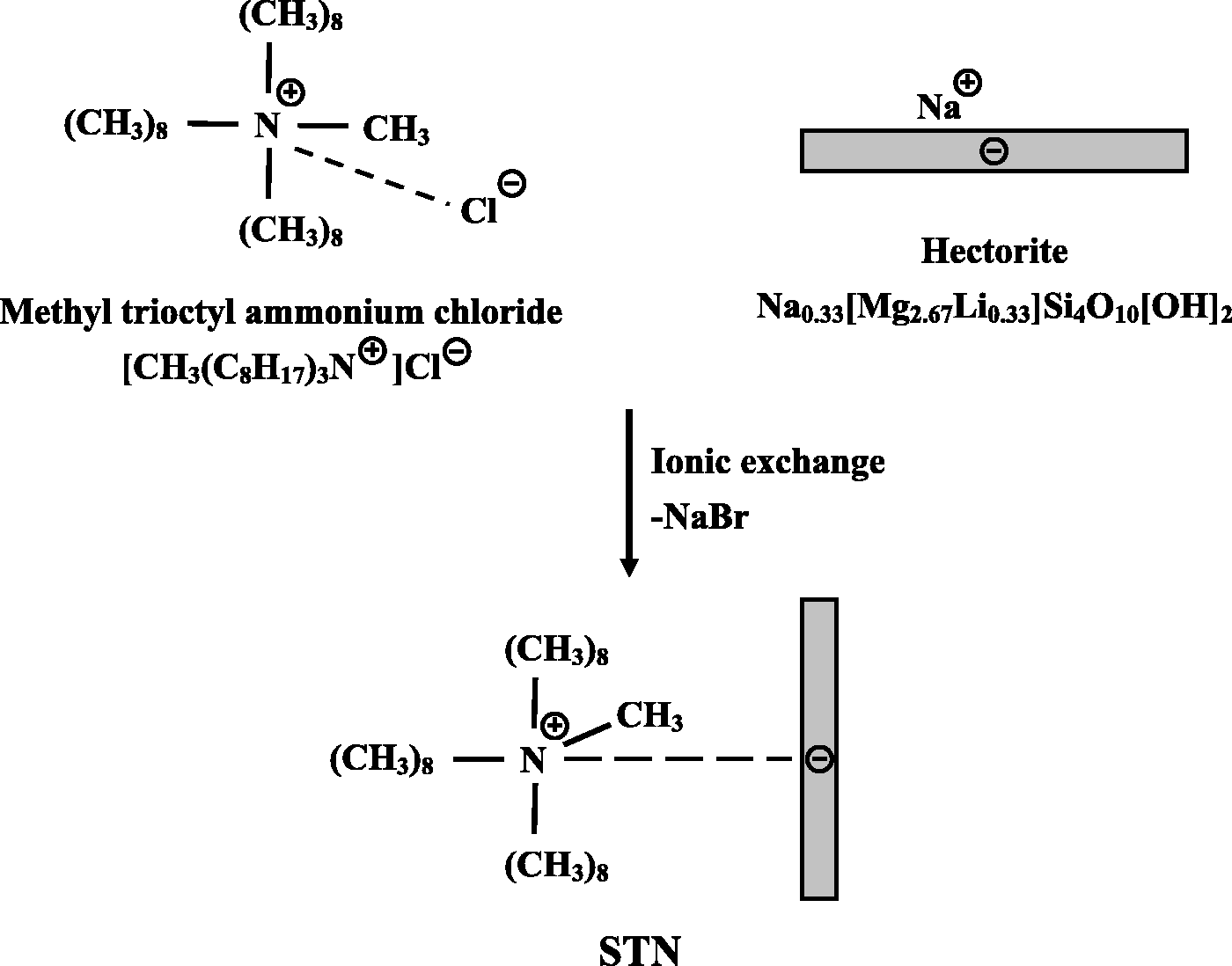
Synthesis of HDA-GSs
Graphene oxide (GO) was synthesized from natural graphite by utilizing a multistep route known as the Hummers method.
33
HDA-GSs were synthesized from HDA and GO according to the following procedure: GO (1 g) was dissolved in 1.5 L of distilled water. HDA (2.00 g; 8.28 × 10−3 mol) was added to 25 mL of ethanol, and the mixture was stirred at 25℃ under a steady stream of N2 and subsequently added to the GO/water mixture. After the mixture was heated for 12 h at 25℃ under a steady stream of N2, it was cooled to 25℃, washed twice with distilled water and ethanol (v/v = 1:1), and dried under vacuum at 70℃ for 24 h in order to obtain HDA-GSs. The synthetic route for HDA-GSs is shown in Scheme 2.
Synthesis of HDA-GS.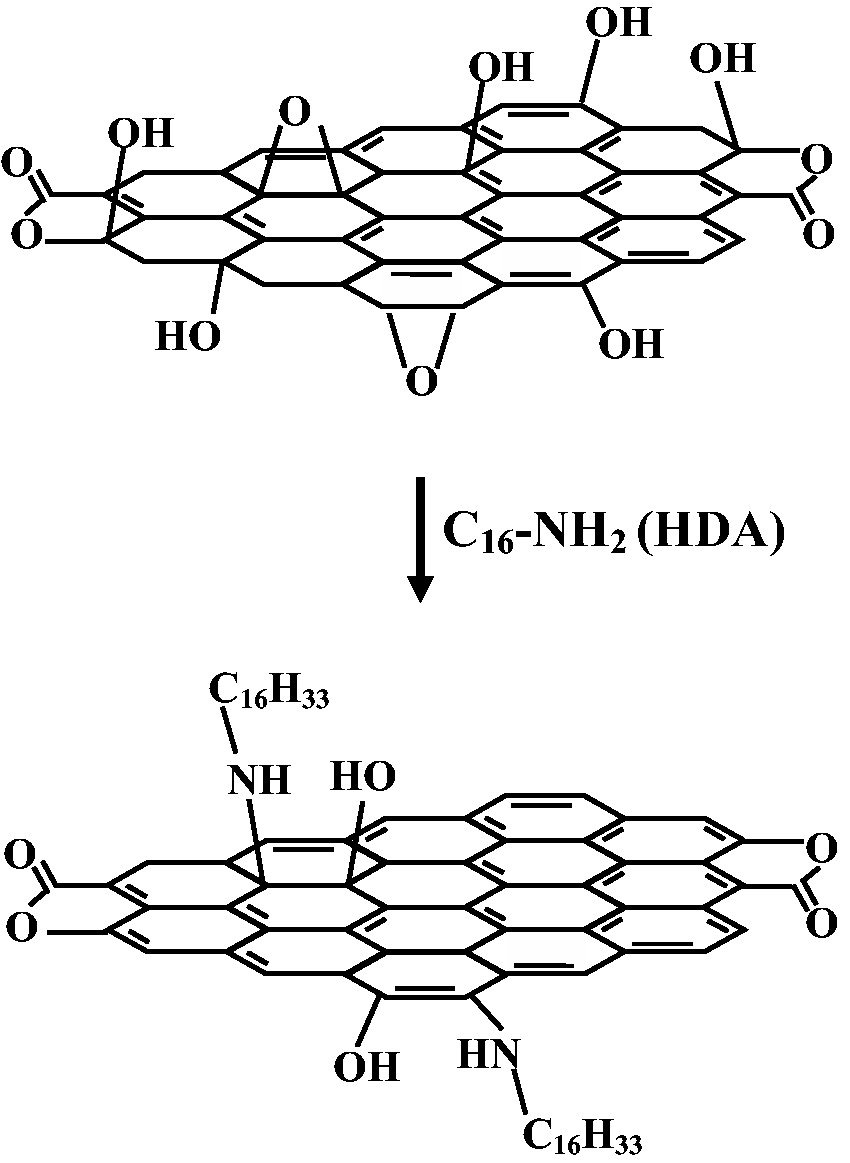
Synthesis of STN/HDA-GS complex
Dried GO (1.0 g) dissolved in distilled water (1.0 L) was added to a 2 L beaker, and stirred at 25℃ for 12 h; 0.5 g of 12-phosphotungstic acid (PTA, H3PW12O40) was suspended in 100 mL of distilled water and placed in a separate flask. It was subsequently added to the GO solution with vigorous stirring for 1 h at 25℃ in order to obtain a homogeneously dispersed solution. The acid cluster of PTA has three protons in the unit structure, which dissociates in the water phase as oxionium ions as H3O+ or H5O2+. Polyanion PTA clusters have been incorporated in hybrid matrices, where they exhibit a strong interaction with the inorganic silica framework and never dissociate from the hybrid matrix.34,35 PTA was therefore used as a binder between the clay and the GSs.
In separate beakers, 2.0 g of HDA and 2.0 g of STN were dispersed in ethanol (100 mL). The STN solution was added dropwise to the aqueous GO solution with vigorous stirring for 30 min in order to obtain a homogeneously dispersed system. Afterward, the HDA solution was added dropwise to the STN/GO solution. After the solvent was removed by filtering, the STN/HDA-GS powder was washed several times with ethanol and dried in a vacuum oven at 80℃ for 12 h.
Preparation of PI hybrid films
The chemical structures relevant to the synthesis of the PI hybrid films are shown in Scheme 3. Poly(amic acid) (PAA) was synthesized from BPA and APS using DMAc as the solvent at 25℃. BPA (5.79 g, 1.96 × 10−2 mol) and DMAc (30 mL) were placed in a three-necked, 250 mL flask. The mixture was then stirred at 0℃ for 30 min under a N2 atmosphere. A solution of APS (4.21 g, 1.96 × 10−2 mol) in DMAc (30 mL) was added to the flask, and the resulting solution was stirred vigorously at 0℃ for 1 h and then at 25℃ for 23 h, yielding a solution of 16.7 wt% PAA in DMAc.
Synthesis of PI nanocomposite films.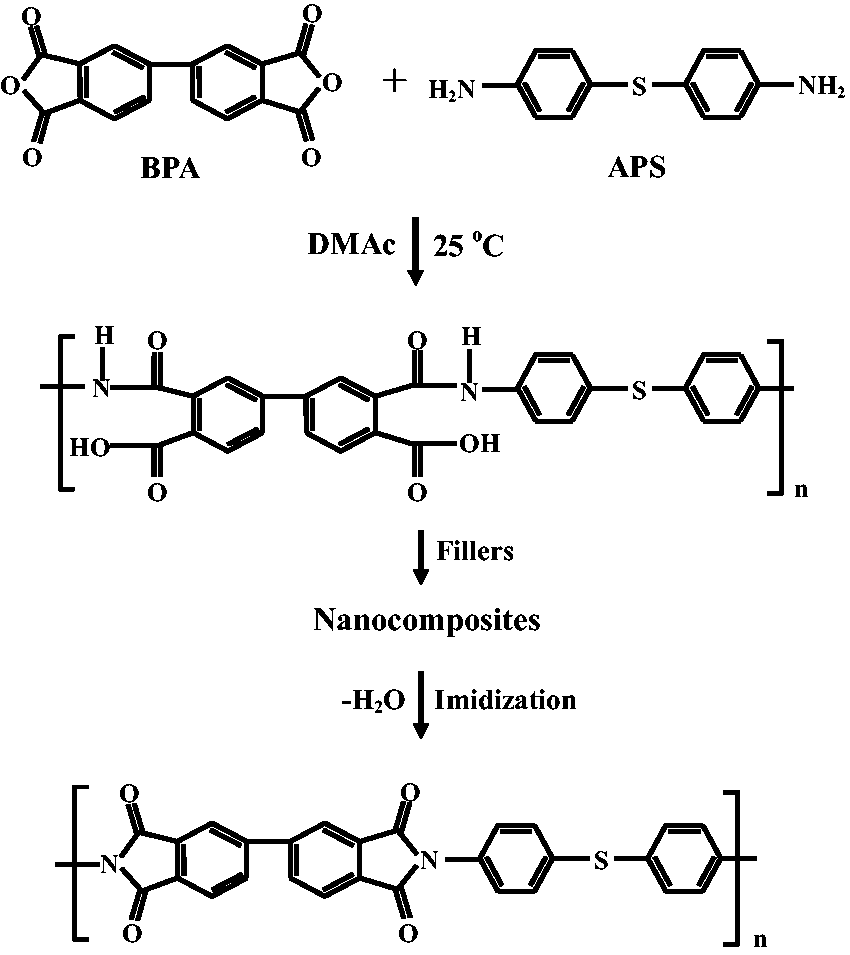
The PI/filler hybrid films were prepared using the solution intercalation method. The synthetic procedures for preparing the hybrid films containing varying amounts of filler are quite similar, therefore a representative procedure (the preparation of the hybrid containing 5 wt% STN) is described here. In a 100-mL beaker, PAA (10 g) was added to DMAc to produce a 12 wt% solution. In a separate beaker, STN (0.52 g) was ultrasonicated in 25 mL of DMAc for 3 h and then added to the PAA solution. The resulting mixture was vigorously stirred for 6 h at 25℃ under a steady stream of N2 to yield a homogeneous dispersion. The resulting solution was cast onto glass plates, and the solvent was evaporated at 50℃ for 1 h. The films were then dried in a vacuum oven at 80℃ for 1 h and then at 110℃ and 140℃ for 30 min each. The PAA film was further imidized on the glass plate through sequential heating at 170, 200, 230, and 250℃ for 30 min at each temperature under a steady stream of N2. The resulting hybrid films were 78–82 µm thick.
Characterization
Fourier transform infrared (FT-IR) spectra were obtained with an FT-IR 460 (JASCO) instrument in the range 4000–600 cm−1 with KBr pellets. A differential scanning calorimeter (DSC, NETZSCH F3) and a thermogravimetric analyzer (TGA, TA Q500) were used at heating rates of 20℃/min under a flow of N2. The coefficients of thermal expansion (CTEs) of the samples were measured using a macroexpansion probe (TMA-2940), which was used to apply an expansion force of 0.1 N to the films at a heating rate of 5℃/min in the temperature range of 50–200℃.
Wide-angle X-ray diffraction (XRD) measurements were performed at room temperature on a Rigaku (D/Max-IIIB) X-ray diffractometer using Ni-filtered Cu Kα radiation. The samples were prepared for transmission electron microscopy (TEM) by placing the PI hybrid films into epoxy capsules and curing the epoxy at 70℃ for 24 h under reduced pressure. The cured epoxies were then microtomed (approximately 90 nm thick), a layer of carbon (approximately 3 nm thick) was deposited on each of the slices; the slices were then placed on copper grids (mesh 200). The TEM images of the ultrathin sections of the PI hybrid films were obtained on a Leo 912 OMEGA TEM using an acceleration voltage of 120 kV.
The O2 permeabilities of the films were measured according to ASTM E96 using a Mocon DL 100. The O2 transmission rates (O2TRs) were obtained at 23℃ and a pressure of 760 Torr (101 kPa).
Result and discussion
FT-IR spectra
The FT-IR spectra of STN, HDA-GS, and STN/HDA-GS are shown in Figure 1.
36
STN was confirmed by FT-IR spectroscopic analysis, as shown in Figure 1(a). FT-IR (K Br): ν (cm−1) 2923 and 2844 (aliphatic –CH2) and 980 (Si–O). The FT-IR spectrum of HDA-GS is shown in Figure 1(b). It contains characteristic absorptions at 3160 (O–H), 2921, 2852 (aliphatic C–H), and 1396 cm−1 (aromatic C–N). The FT-IR spectrum of STN/HDA-GS is shown in Figure 1(c). It contains characteristic absorptions at 2905, 2855 (aliphatic C–H), 1397 (aromatic C–N), and 1024 cm−1 (Si–O).
FT-IR spectra of STN, HDA-GS, and STN/HDA-GS.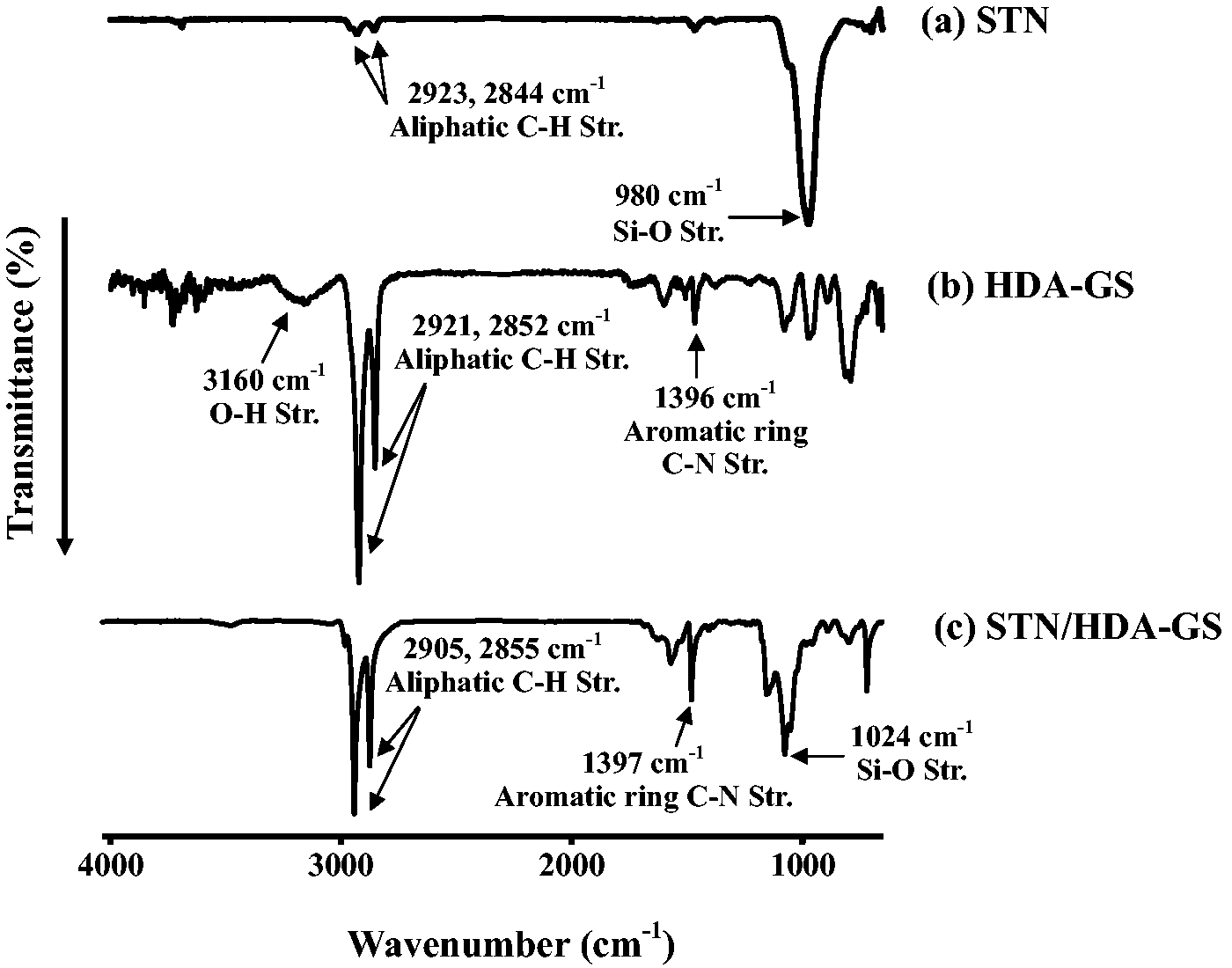
The formation of PAA and the completion of imide formation from amic acid were also confirmed by examining the FT-IR spectra; the spectra of PAA and the PIs are shown in Figure 2. PAA exhibits a broad absorption band at around 2800–3400 cm−1 because of the acid (O–H) group of PAA. The C = O stretching peaks at 1711 and 1590 cm−1 are due to the acid and amide groups of PAA, respectively, and they shift to higher wavenumbers in the imides, specifically to approximately 1775 cm−1 (C = O, in phase) and 1717 cm−1 (C = O, out of phase), respectively. In addition, the presence of a peak at 1356 cm−1 corresponding to C–N–C stretching confirms the formation of the imides.
36
FT-IR spectra of poly(amic acid) (PAA) and polyimide (PI).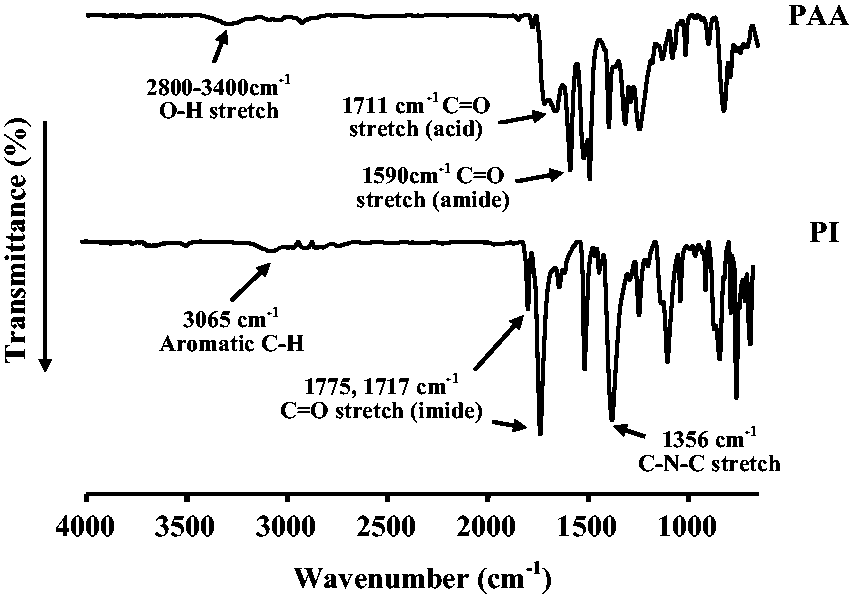
Dispersion
Figure 3(a) displays the XRD patterns of STN at 2θ between 2° and 10°. The reflection of STN was found at 2θ = 4.36° [interlayer spacing (d) of 20.22 Å]. Figure 3(a) also displays the XRD curves for pure PI and the PI hybrid films with varying STN contents in the range of 0 to 20 wt%. When the PI hybrid films containing up to 5 wt% STN were formed, the organoclay peak at 2θ = 4.36° disappeared from the diffraction pattern. This result indicates that the clay layers were exfoliated and homogeneously dispersed throughout the PI matrix, which provides supplementary evidence that the STN/PI hybrids were nanocomposites.37,38 The intensity of the XRD peak at 2θ = 4.73° (d = 18.68 Å), however, formed suddenly as the clay loading increased from 5 to 10 wt%, suggesting that the dispersion was more effective at lower clay loadings than at higher clay loadings. Higher clay loadings were expected to increase the aggregation in some portions of the clay within the PI matrix. As shown in Figure 3(a), the hybrid films containing 10 and 20 wt% of STN exhibit a diffraction peak at 2θ = 4.73° and 4.88° (d = 18.68 and 18.09 Å, respectively). This peak shifted slightly relative to that of STN, which was observed at 2θ = 4.36° (d = 20.22 Å). This result revealed that some parts of the clay collapsed within the PI matrix. One hypothesis is that thermal decomposition of the organic portion of STN collapsed the clay layers, thus reducing the d-spacing.
39
Yano et al.
40
also reported that weakly bound organic molecules can detach from the clay surface. This detachment, which is caused by the heat treatment during the cyclization of PAA to PI, leads to a reduction in the interlayer spacing relative to that of STN. Substantial increases in the intensities of the XRD peaks were observed when the clay loading was increased from 10 to 20 wt%, which suggests that the clay easily aggregates at higher amounts of clay loading.
XRD patterns of pure fillers and PI hybrids with various filler contents.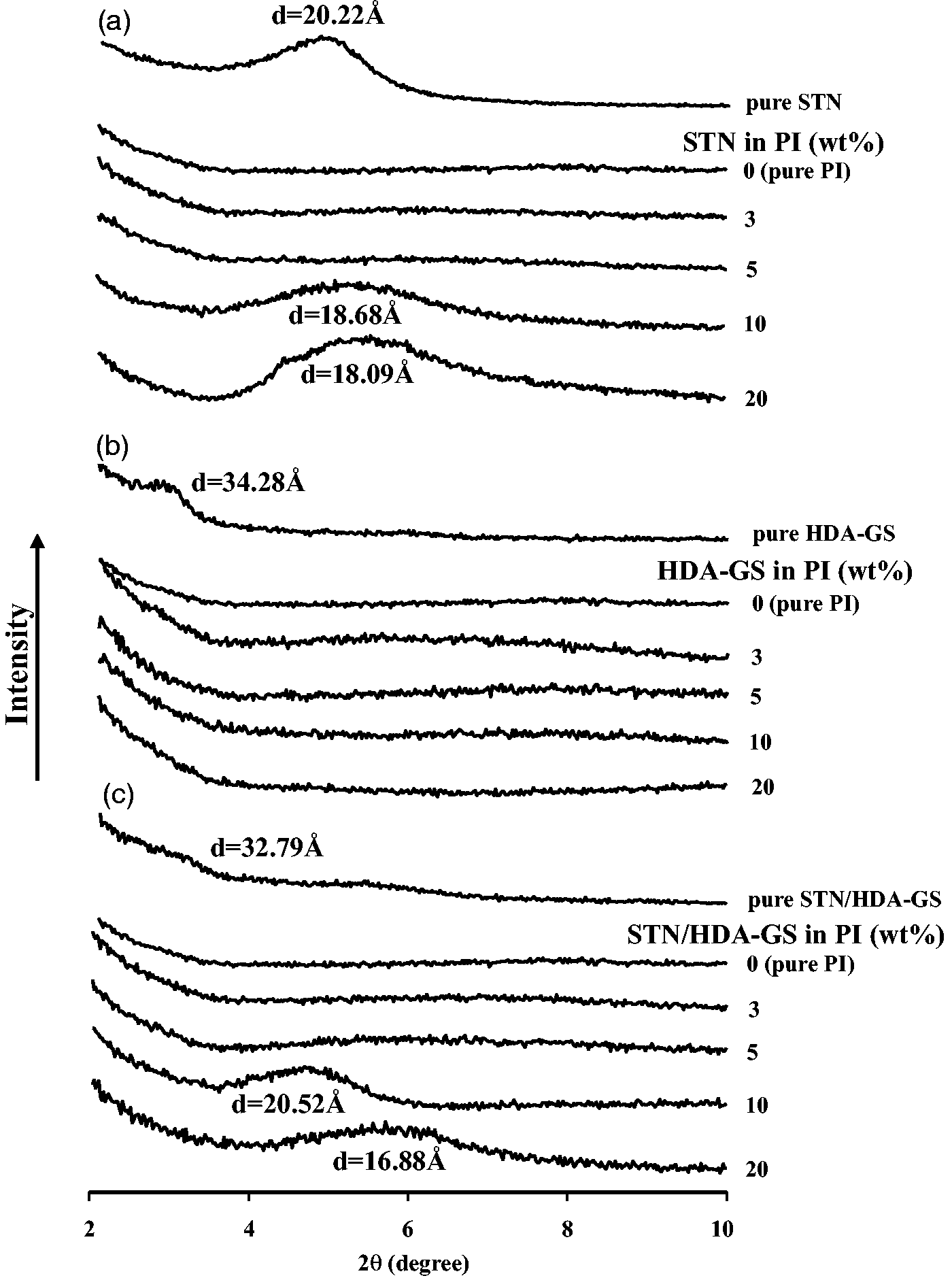
The XRD traces of the pure HDA-GS and HDA-GS/PI hybrid films are shown in Figure 3(b). The d001 reflection for HDA-GS occurred at 2θ = 2.57°, which corresponds to an interlayer spacing of 34.28 Å. The GS peak disappeared from the XRD curves of the PI hybrid films with 3–20 wt% HDA-GS, which indicates that the GSs were exfoliated and homogeneously dispersed throughout the PI matrix. XRD offers a convenient method to determine the interlayer spacing as a result of the periodic arrangement of the graphite layers in the pristine GSs and in the intercalated hybrids. Although XRD enables precise routine measurements of the GS layer spacing, it does not enable the spatial distributions of GSs to be determined nor does it enable any inhomogeneous sections of the hybrids to be detected. Additionally, some layered GSs do not initially exhibit well-defined basal reflections, thus peak broadening and reductions in intensity are difficult to follow systematically. Therefore, any conclusions about the mechanisms of the hybrid formation and microstructure based solely on XRD results are only tentative.41,42 Further evidence of the GS dispersion in the PI films on a nanoscale was obtained with TEM, as described in the morphology section.
The XRD traces of the PI hybrids with varying STN/HDA-GS contents are shown in Figure 3(c). The d001 reflection for STN/HDA-GS occurred at 2θ = 2.69°, which corresponds to an interlayer spacing of 32.79 Å. This reflection peak is probably derived from HDA-GS, as shown in Figure 3(b). For the PI hybrids containing 0–5 wt% STN/HDA-GS, no obvious filler peaks were present in the XRD curves. For PI containing a 10 wt% STN/HDA-GS, however, a medium peak at d = 20.52 Å (2θ = 4.30°) existed in the XRD trace for the hybrid film. Substantial reductions in the interlayer d-spacings of the XRD peaks were observed for STN/HDA-GS loadings from 10 to 20 wt% (2θ = 5.23° and d = 16.88 Å). The reduction in the interlayer spacing from 20.52 Å to 16.88 Å was caused by the detachment of organic molecules from the organically modified nanofiller (STN and FGS) surface upon heat treatment, which occurs during the cyclization process from PAA to PI. This squeezing mechanism should also serve to inhibit the exfoliation of the nanofiller in the PI, causing the filler to have an intercalated structure.
Morphology
XRD is by far the simplest method available for measuring the interlayer spacing of these hybrid materials. However, we extended this analysis by using TEM to evaluate the degree of intercalation and the amount of aggregation of the filler clusters. Additional direct evidence for the formation of a true nanocomposite can be provided by TEM analysis of ultramicrotomed sections. Figures 4 and 5 display the micrographs of the PI hybrid films with STN and HDA-GS, respectively. The areas marked by the arrows in (a) are shown enlarged in (b). The dark lines in the photographs are the intersections between the clay (which was 1 nm thick) and the GSs, whereas the distance between the dark lines is the interlayer distance.
TEM micrographs of PI hybrid film containing 5 and 20 wt% STN with increasing magnification levels from (a) to (b). TEM micrographs of PI hybrid film containing 5 and 20 wt% HDA-GS with increasing magnification levels from (a) to (b).

Figure 4 displays the morphology of a PI hybrid containing 5 wt% STN. In this sample, each layer of the polymer matrix consisted of well-dispersed individual clay layers (dark lines), and some of the clay aggregated to thicknesses of approximately 5 nm. As was the case for the hybrid material with 20 wt% STN (see Figure 4), the clay was mostly aggregated in the polymer matrix; however, the average particle size was found to be below 20 nm, as calculated from the TEM photographs. The presence of a peak with a high intensity in the XRD patterns of these samples can be attributed to the aggregated layers (see Figure 3a). The XRD and TEM results indicate that the clay particles at a lower clay content were more dispersed than those at a higher clay content throughout the PI matrix.
Typical TEM images of the PI hybrid films containing 5 and 20 wt% HDA-GS are shown in Figure 5. Figure 5 shows that the GSs were well dispersed within the polymer matrix at all magnification levels. The GS layers of the HDA-GS hybrids were exfoliated within the matrix polymer, unlike the hybrids containing STN, which was also confirmed by XRD, as shown in Figure 3(b).
A different type of behavior was observed for the PI hybrid films with the STN/HDA-GS complex. The chemical structure and TEM micrograph of the STN/HDA-GS complex are shown in Figure 6. Interestingly, Geim et al.
43
reported that graphene forms straight rigid platelets in the composite, indicating that the graphene is extremely stiff. These samples in the current study exhibited platelet morphology with uniform orientation distribution, as confirmed by area (2) in the TEM micrograph shown in Figure 6.
TEM micrograph of the clay/GS complex: (1) clay and (2) graphene sheet.
Figure 7 displays the morphology of a PI hybrid containing 5 and 20 wt% STN/HDA-GS. These fillers were mostly aggregated within the polymer matrix, but the average particle size was found to be less than 100 nm, as calculated from the TEM photographs. The complex filler layers of the 20 wt% STN/HDA-GS were highly aggregated, unlike the hybrids containing 5 wt% STN/HDA-GS, which is consistent with the XRD data shown in Figure 3(c). These findings suggest that during the intercalative polymerization process, bonded double-layered clay/GS structures (see Figure 6) inhibited polymer chain interpenetration between filler layers and subsequently aggregated within the polymer matrix. The laminated filler sheets were laterally aggregated with one another, and various bundles of fillers were formed and stacked.
TEM micrographs of PI hybrid film containing 5 and 20 wt% STN/HDA-GS complex with increasing magnification levels from (a) to (b).
The XRD and TEM results indicate that the fillers were well dispersed throughout the PI matrix at low filler contents, whereas aggregated structures were present at higher filler contents and that the dispersion of STN and HDA-GS was better than that of STN/HDA-GS in the PI matrix. The clay/GS complex layers of the PI hybrids were aggregated within the matrix polymer, unlike the hybrids containing STN and GS. The unusual thermal and gas barrier properties of these hybrid films are discussed in the following sections with respect to the dispersion of the nanofillers.
Thermal properties
Thermal properties of polyimide (PI) hybrid films with various filler contents.

HAD-GS: hexadecylamine-graphene sheet.
At a 2% initial weight-loss temperature.
Weight percent of residue at 600℃.
Coefficient of thermal expansion for 2nd heating is 50–200℃.
The thermal stabilities of pure PI and the PI hybrids are also listed in Table 1. The initial degradation temperatures (TDi) of the hybrids at 2% weight loss were found to gradually decrease as the filler content increased to 20 wt%. For example, TDi varied from 503℃ to 424℃ upon increasing the STN content of the hybrids from 0 to 20 wt%, and the largest decrease of 79℃ arose in the case of 20 wt% STN relative to that of pure PI. Similar results were observed in the samples containing HDA-GS and the STN/HDA-GS complex, as shown in Table 1.
It is generally believed that the exfoliation of clay and graphene components in polymer materials can improve the thermal stability of the polymers based on the fact that these species possess desirable thermal stabilities. The addition of these fillers also enhances TDis since they act as insulators and mass-transport barriers to the volatile products generated during decomposition, as shown in Scheme 4.47,48 In our investigation, however, the thermal stabilities of composites containing STN, HDA-GS, and STN/HDA-GS complexes were found to gradually decrease as the filler content increased up to 20 wt%. These low values of TDis were most likely a result of the organically modified nanofillers being thermally unstable. The weight of the residue at 600℃ (wtR600) was virtually unchanged when the filler loading was varied; specifically, it increased from 80% to 84% for STN, from 80% to 83% for HDA-GS, and from 82% to 85% for STN/HDA-GS.
Various structures of nanofillers in matrix polymer.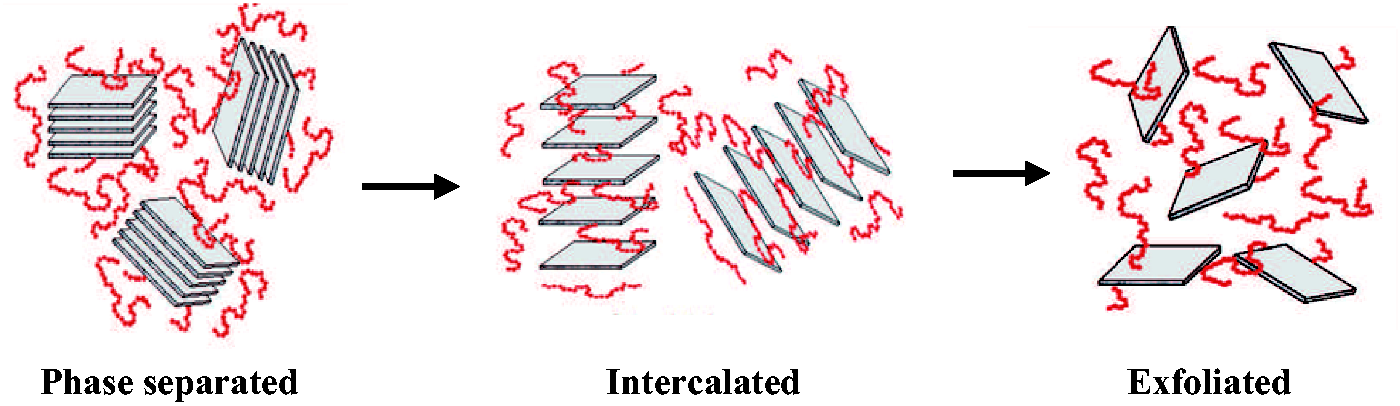
The CTE values for the PI hybrids with different types of nanofillers in the temperature range of 50–200℃ are also listed in Table 1. The CTE of the STN hybrid films exhibited a trend that was quite different from those of Tg and TDi, i.e. the CTE decreased as the filler content increased to 20 wt%. The lowest CTE value for the STN hybrid films (i.e. 31.81 ppm/℃) was observed at a loading of 20 wt%. The CTEs of the HDA-GS hybrids also decreased significantly as the GS loading increased to 20 wt%. For example, the CTEs of the PI hybrids decreased from 60.49 to 6.91 ppm/℃ when the GS loading increased from 0 to 20 wt%. This result implies that the magnitude of the reduction in thermal expansion is a result of the graphene layers depending on the orientation of the PI molecules and the rigid nature of the graphene layers. Upon heating, in-plane-oriented PI molecules tend to relax in a direction normal to their original direction, and they subsequently expand in the out-of-plane direction. 49 The graphene layers are much more rigid than the PI molecules and do not deform or relax as easily as the PI molecules. As a result, the graphene layers inhibit the thermal expansion of the PI molecules very effectively in the out-of-plane direction.
For the STN/HDA-GS hybrid, the CTEs of the PI hybrids decreased from 60.49 to 42.76 ppm/℃ when the amount of STN/HDA-GS filler increased from 0 to 20 wt%, which is the least desirable result among the three types of fillers. It appears that the clay/graphene double-layer structures (see Figure 6) may inhibit the effective dispersion of the filler within the PI matrix and diminish its insulation effect, thus deteriorating the CTE value of the hybrid film. These poor results for the CTE also seem to be caused by filler aggregation, as confirmed by the TEM micrograph (Figure 7).
Gas permeation
In general, the gas permeabilities of the hybrid films decreased when the inorganic component increased. In organic polymers filled with exfoliated inorganic materials, the gas permeability can decrease with respect to the polymer-filler adhesion and compatibility. 50 An exfoliated filler that possesses desirable compatibility with the organic polymer matrix typically decreases the permeability, which is a result of the reduction in the transport cross section and an increase in the lengths of the tortuous paths for the gas molecules, as shown in Scheme 4.51,52
Barrier properties of polyimide (PI) hybrid films with various filler contents.
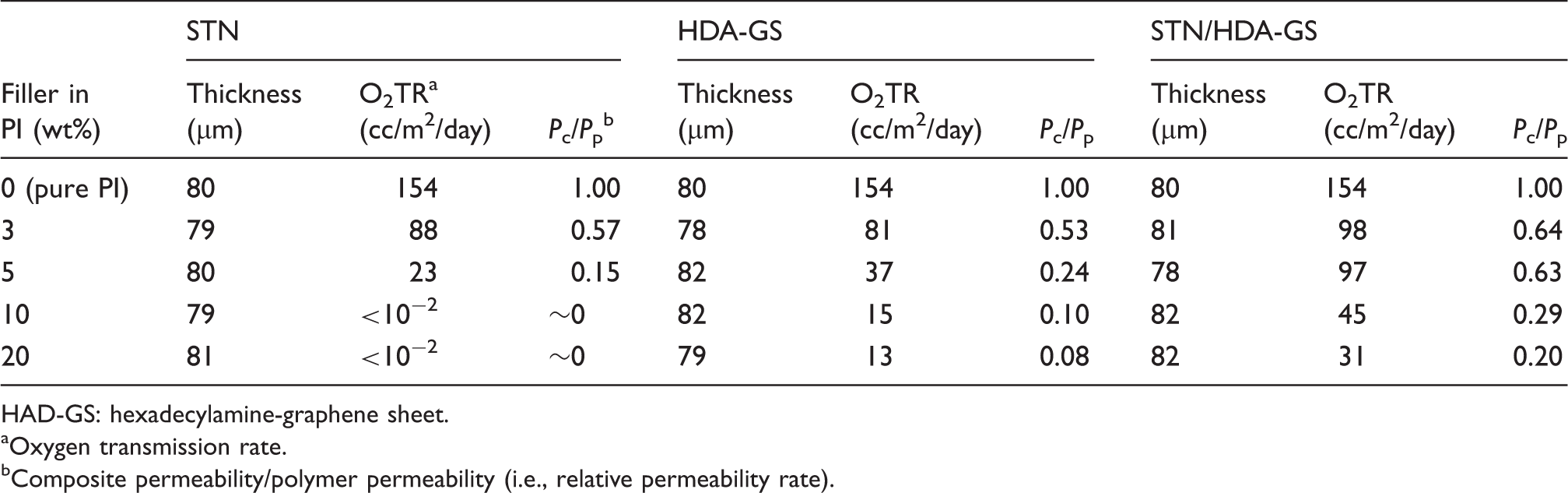
HAD-GS: hexadecylamine-graphene sheet.
Oxygen transmission rate.
Composite permeability/polymer permeability (i.e., relative permeability rate).
The HDA-GS hybrid films exhibited lower O2 permeability values than the pure PI film. It was also determined that the permeability of O2 was influenced by the HDA-GS content within the polymer matrix. With the HDA-GS loading increasing from 3 to 20 wt%, the permeability values decreased linearly from 81 to 13 cc/m2/day, which is a result of an increase in the lengths of the tortuous paths for O2 molecules. It also appears that the films containing higher amounts of HDA-GS were much more rigid, which contributed to the decrease in gas permeability.
The O2TRs for the STN/HDA-GS hybrid films with clay contents between 3 and 20 wt% are also summarized in Table 2, and similar results were obtained. These results confirm that the gas transfer process for this penetrant is highly dependent on the filler loading level. For example, the addition of 20 wt% of STN/HDA-GS complex (31 cc/m2/day) resulted in an 80% reduction in the permeability rate of O2 relative to that of the pure PI film (154 cc/m2/day); i.e. the permeability values of the STN/HDA-GS hybrids are about one-fifth that of pure PI.
It is thus demonstrated that the gas barrier effect of STN is the best among the three types of nanofillers with respect to the O2TR. The presence of clay introduces a tortuous path for a diffusing penetrant. The reduction in permeability arises from the longer diffusive path that the penetrants must travel in the presence of the clay filler. The sheet-like morphology is particularly efficient in maximizing the path length as a result of the large length-to-width (L/W) ratio relative to shapes of fillers54,55 such as spheres or cubes.56,57 This reduction in permeability is primarily attributed to the fine dispersion (exfoliation) of the clay particles, up to 20 wt% clay content in the PI matrix, as shown in Figure 4. However, in contrast to STN, no significant changes in O2TR were observed for the STN/HDA-GS complex as a result of the aggregation of fillers in PI, as shown in Figure 7.
Conclusions
We have investigated the dispersibility of various types of nanofillers in PI, with the aim of improving the various properties of the PI hybrid films. STN, HDA-GS, and an STN/HDA-GS complex were tested in the PI hybrids. The PI hybrid films with varying filler contents ranging from 0 to 20 wt% were synthesized using the solution intercalation method. The thermal properties, morphology, and gas permeability of the PI hybrid films were then compared in detail. Our results confirm that these properties are dependent on the types of nanofillers incorporated in the polymer and the amount present within the PI polymer matrix.
The morphologies of the hybrids were examined by TEM, and it was confirmed that STN and HDA-GS have better dispersion properties than the STN/HDA-GS complex with respect to the PI matrix. This observation is in agreement with the variations in the thermal properties and gas barriers of these hybrid materials with the same filler loading levels within the hybrid films.
Although both STN and HDA-GS exhibit desirable dispersion throughout the PI matrix, the Tg and TDi values of the STN hybrids are superior to those of the HDA-GS hybrids. However, the CTE of the HDA-GS hybrids is superior to the CTE of the STN hybrids. The gas barrier effects of STN are greater than those of HDA-GS or the STN/HDA-GS complex. Furthermore, it was determined that adding only small amounts of fillers were sufficient to improve the properties of PI.
Before we initiated this study, we expected to obtain ideal properties from the STN/HDA-GS hybrids, which contained both clay and graphene. However, no such results were observed from the STN/HDA-GS complex hybrids as a result of the double-layered complex structure of clay and graphene on the interlayer bonding.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.