
Abstract
High thermal conductivity is one important factor in the selection or development of ceramics or composite materials. Predicting the thermal conductivity would be useful to the design and application of such materials. In this paper, a multi-scale model is developed to predict the effective thermal conductivity in SiC particle-reinforced aluminum metal matrix composite. A coupled two-temperature molecular dynamics model is used to calculate the thermal conductivity of the Al/SiC interface. The electronic effects on the interfacial thermal conductivity are studied. A homogenized finite element model with embedded thin interfacial elements is used to predict the properties of bulk materials, considering the microstructure. The effects of temperatures, SiC particle sizes, and volume fractions on the thermal conductivity are also studied. A good agreement is found between prediction results and experimental measurements. The successful prediction of thermal conductivity could help a better understanding and an improvement of thermal transport within composites and ceramics.
Introduction
Due to their excellent properties including high strength-to-weight ratio, good wear resistance, and high strength retained at elevated temperatures, ceramics-reinforced metal matrix composites are desired for many critical components in structures, engineering systems, and medical applications. Among these materials, silicon carbide (SiC) reinforcement is widely used due to its high thermal conductivity for various semiconductor devices, capsule materials for nuclear equipment, and gas seal rings in compressor pump.
Extensive experimental studies have been conducted to measure the thermal conductivity of SiC ceramics or SiC-reinforced metal matrix composites for different microstructure.1–6 Considering the wide range of material compositions under various conditions, a systematic understanding of thermal transport is needed for SiC-reinforced metal matrix composites to reduce the cycle of trial-and-error methods. To this end, it is necessary to develop a predictive model to accurately calculate the thermal conductivity for various compositions at different temperatures. As the interface greatly influences the thermal conductivity, an appropriate way of calculating interfacial thermal conductivity is needed. One approach that has been widely used is the Hasselman and Johnson 7 model. Hasselman and coworkers6,7 considered the interfacial conductance between different constituents and proposed an analytical solution for the effective conductivity of particles-reinforced composites. Extensive studies have used the Hasselman–Johnson model to evaluate the effect of interfacial thermal conductance. With an assumed range of interfacial conductance, the effective thermal conductivity of SiCf/SiC composites was studied. 8 The Hasselman–Johnson model can further be used to calculate the interfacial conductance by fitting it to experimental results. By varying the size of the reinforcement particles,6,9 the interfacial thermal conductance of particle-reinforced metal matrix composites can be calculated using the Hasselman–Johnson 7 model. Using the derived inverse equation, Nan et al. 10 calculated the interfacial thermal resistance from effective thermal conductivities, despite the implementation of calculation equations different from the Hasselman–Johnson model. Using finite element methods (FEM) modeling, Islam and Pramila 11 studied the effect of interfacial thermal conductance on the thermal conductivities of fiber-reinforced composites, where the interfacial thermal conductance was obtained by curve-fitting of experimental results.
Despite the convenience of the Hasselman–Johnson model in evaluating interfacial thermal conductance, it provides no understanding of interfacial thermal transport, such as temperature gradients, the size of interfacial region, and interfacial thermal conductivity. Additionally, the calculation accuracy largely depends on experimental measurements. Hence, a numerical method is needed to directly simulate the interfacial thermal transport and calculate the interfacial thermal conductivity.
Several recent studies have dealt with the interfacial thermal conductance by the molecular dynamics (MD) method.12–16 Depending on whether the simulations are for conductivity under constant temperature or not, classical MD simulations can be classified into equilibrium molecular dynamics (EMD) and nonequilibrium molecular dynamics (NEMD) simulations. A well-known method of calculating the thermal conductivity of materials based on the atomic trajectories from EMD simulations is the Green–Kubo formula,17,18 which correlates thermal conductivity to the heat current autocorrelation function. Different from EMD simulations, NEMD simulations are realized by imposing a heat flux or a temperature difference across a material system.17–19 In NEMD simulations, the temperatures can be controlled by adding energy or rescaling temperatures of atoms. With the temperature gradient
While MD simulations can model thermal transport, most of studies are focused on the phononic contribution due to the incapability of MD simulations in explicitly modeling the contribution of electrons. The two-temperature model (TTM) has been introduced to include electron–phonon coupling. The electron and phonon are described as two separate subsystems with an assigned temperature for each. To incorporate electronic effects into the modeling of radiation damage simulations, Duffy and Rutherford22,23 introduced a coupled TTM-MD scheme to account for electron–phonon coupling, electronic stopping, and both the temporal and spatial evolution of the phononic and electronic subsystems. Based on Duffy and Rutherford’s scheme, Phillips and Crozier 24 added a communication process between the electronic and atomic subsystems and achieved energy-conserving TTM-MD simulations. Wang et al. 13 extended the conventional NEMD simulation by coupling it with the TTM model to analyze the interfacial thermal transport across two different metal/nonmetal interfaces. A better agreement with experimental data was obtained compared with conventional NEMD simulations with only phonons considered. Using a coupled TTM-MD scheme, Jones et al. 14 studied the size and electronic effects on the thermal conductance of Al/GaN interface. A linear relationship was found between the interfacial resistance and inverse system length while electron transport had little effect on the interfacial thermal conductance, which is also supported by the studies on several other metal/nonmetal interfaces. 15 To reliably predict the thermal conductivity of SiC particle-reinforced aluminum matrix composites, the TTM-MD simulations were implemented in this study to evaluate the electronic contributions to the thermal transport of Al/SiC interface.
Despite the convenience of MD simulations in predicting thermal properties, the empirical interatomic potentials may not be available for MD simulations, which necessitates the need of ab initio calculations25,26 to predict the interatomic potentials. Additionally, it is limited by the simulation system size and computation costs for bulk materials. Finite element (FE) modeling provides a convenient way to evaluate the effective thermal conductivity of bulk materials through homogenization of one representative volume element (RVE).27–29 To fully take advantage of ab initio calculations, MD simulation, and FE analysis, it is necessary to develop a multi-scale model that can incorporate the benefits of calculations at each scale.
In this study, a multi-scale model is developed to predict the thermal conductivity of SiC particle-reinforced aluminum matrix composites by bridging ab initio calculations, MD simulations, and FEM. The interatomic potentials are derived by the ab initio method and subsequently used in MD simulations. The multi-scale model implements the temperature-dependent interfacial thermal conductivity calculated from TTM-MD simulations, where electronic effects are explicitly evaluated. The actual microstructures of composites are considered by varying SiC particle sizes and volume fractions in the FE analysis. The developed multi-scale model can not only implement the benefits of ab initio calculations and MD simulations in considering material microstructure at small scale and predicting interfacial thermal conductivity but also greatly reduces computation costs through FE analysis.
Multi-scale modeling of thermal conductivity
The derivation and selection of interatomic potentials
At different levels of temperatures, SiC particle-reinforced aluminum metal matrix composites of varying particle sizes and volume fractions are studied. To calculate material properties in MD simulations, it is very important to use accurate interatomic potential models. Without adding any additional element in this type of composites, only two constituents, i.e. SiC and Al, exist and hence, the interface between SiC particle and Al matrix is a mixture of SiC and Al.
Since little information is available for the measured potential data of typical elements in SiC-reinforced aluminum metal matrix composites, i.e. Al-Si/C in this study, interatomic potentials need to be determined using a predictive method. Ab initio methods provide a convenient way of obtaining interatomic potentials. A predefined interatomic potential based on density functional theory is usually used with curve-fitted parameters.30,31 By fitting the total energy of interatomic potentials to the results by ab initio calculation, the parameters of the interatomic potential model can be determined.
To calculate the interatomic potentials, the first step is to get the adhesive energies of the established system as the source data. Then, an analytical formula can be derived by using the inversion method,
32
which expresses the interatomic potential in terms of adhesive energies. It is assumed that the adhesive energy is equal to the sum of interactions across the interface, which is a first-order but practical approximation for the metal/semiconductor interface. Because the charge transfer at the interface is mainly confined to the first Al and SiC layers,
33
the three-body interaction j–i–k is limited in this region, which means the central atom i is located in the first SiC layer, and j, k are chosen from the neighboring Al and SiC layers. For the three-body interactions, the modified Stillinger–Weber (MSW) potential form has been widely used in covalent matters with zinc blende structures, which is the typical interface structure of SiC ceramics. The MSW form describing three-body interaction is shown as
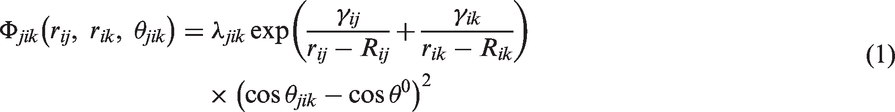
Based on the interatomic potential derivation, ab initio calculation of adhesive energy of the interface structure was carried out using a CASTEP program based on the Al/SiC interface structure. A CASTEP program with generalized gradient approximation was used to calculate the adhesive energy of the interface structure, where a plane-wave cutoff energy of 400 eV was used with a K-point mesh of 7 × 7 × 3. To validate the accuracy of the ab initio calculation, the interatomic potentials of Al-Si and Al-C pairs were calculated in order to compare with the ab initio calculation results of Al-SiC (100) interface in literature.
32
As can be seen from the comparison in Figure 1, the calculated interatomic potential results agree very well with the data obtained in the literature.
Interatomic potentials comparison between results by Zhao and Chen
32
and ab initio validation results.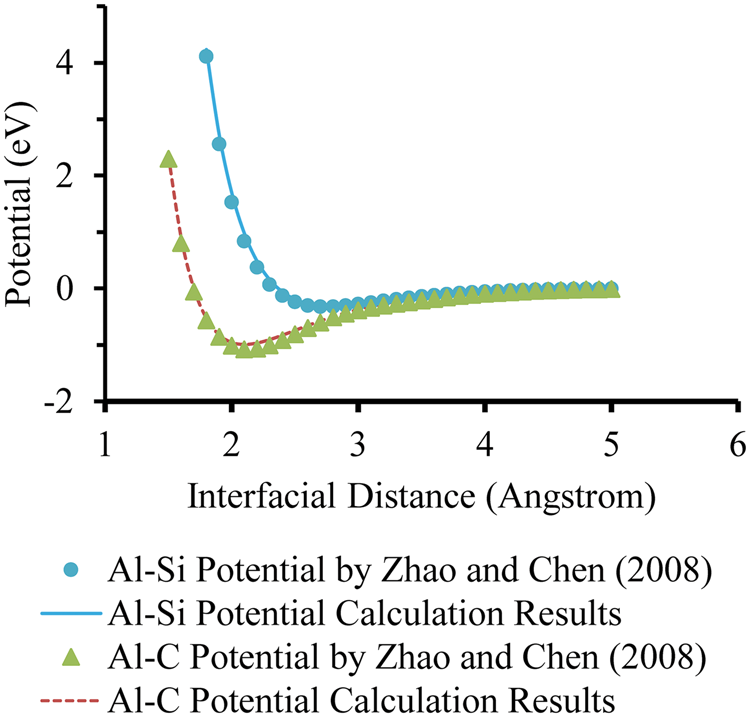

Morse potential function parameters parameterized to the ab initio data.

Stillinger–Weber potential parameters for the Al/SiC interface.

SiC: silicon carbide.
The embedded-atom method (EAM) as shown in equation (3) was used to model aluminum in the MD simulations through a summation of pairwise interaction and the energy required to embed an atom in the electron density produced by the local site.
34

EAM potential parameters for aluminum.
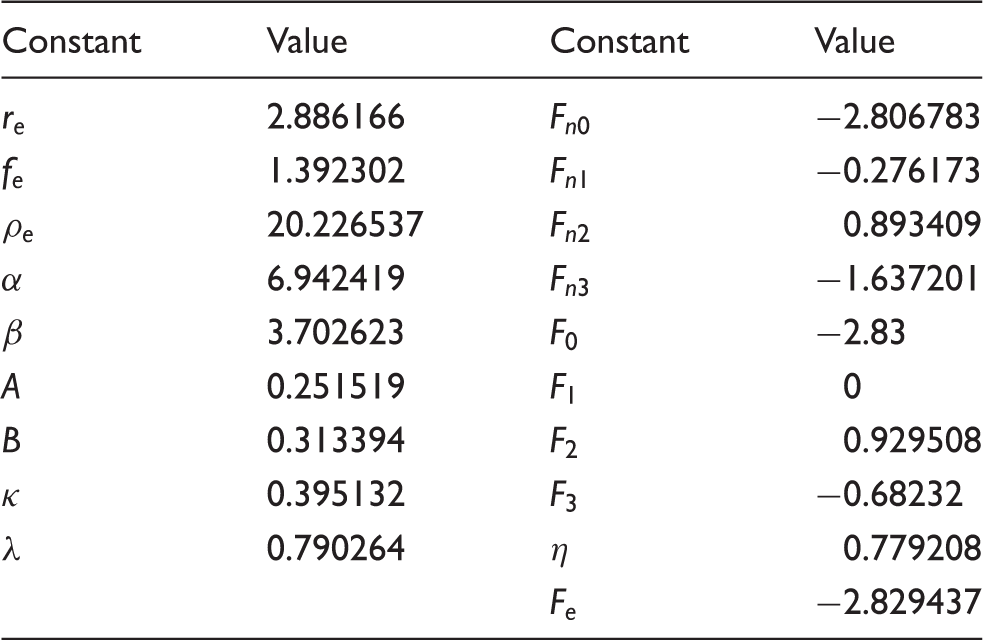
EAM: embedded-atom method.
Tersoff potential parameters for silicon carbide.
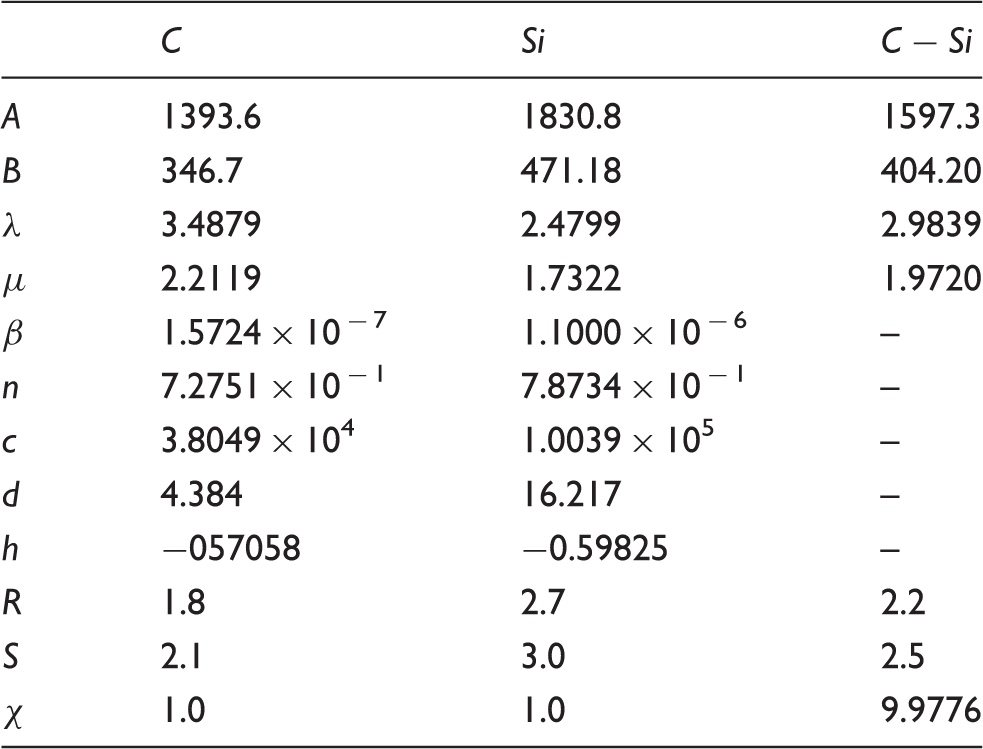
MD equations of thermal conductivity in solid materials
The RNEMD algorithm comprises two components: a means of moving energy from the cold to the hot slab and a way of calculating the temperature gradient. For the calculation of thermal conductivity, the numerical system of heat conduction in a solid material is considered as shown in Figure 2. In the heating and cooling layers, the atoms are heated and cooled, respectively, and the heat is conducted through the atoms between them, i.e. conduction layer.
Configuration of heat conduction layers.
38
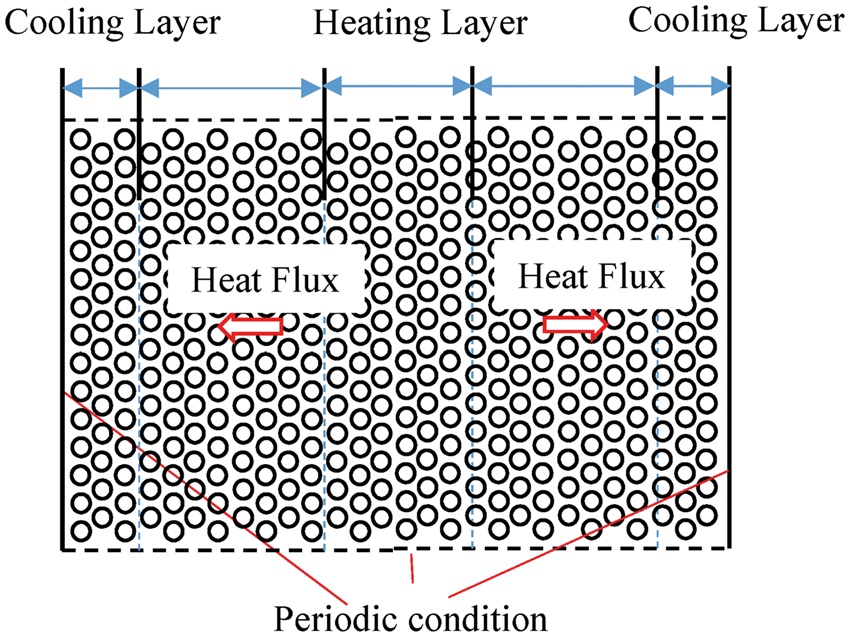
The heat flux is given by the sum of all energy transfer per time per area

To calculate the thermal conductivity, the second task is the calculation of the temperature gradient. The temperature T within each layer can be evaluated by



A linear response in the material system could be assumed as can be seen for all cases17,18,20,39 in the MD calculation of thermal conductivities. Hence, the temperature gradient in the heat conduction direction can be assumed as

Combining equations (4)–(8) with the Fourier’s law, an approximation of thermal conductivity can be obtained as
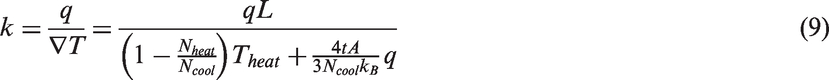
In equation (9), L and A are related to the dimensions of the material system, and t is determined by the simulation time. These parameters are pre-determined and remain the same once the simulation setup is fixed.
In order to induce heat conduction through the atom layers, there are two types of thermal systems for the heating and cooling layers: constant temperature and constant heat flux. In the system of constant temperature, the heating and cooling layers are kept at constant high and low temperatures, respectively, whereas the constant flux system maintains a constant heat flux at the heating layer and subtracts the heat flux of the same amount from the cooling layer. In the current study, the latter method is employed to minimize computation time since the calculation of heat fluxes takes much time in the averaging manipulation. As can be seen in equation (9), after the heat flux q is given, the thermal conductivity k is determined by
The initial velocities of the simulation system are determined from the desired temperature for thermal conductivity calculation. The periodic boundary conditions are applied to all directions of the simulation system. The heat flux is added and subtracted by rescaling the kinetic energy of the heating and cooling layers, respectively. A constant-pressure-and-temperature ensemble first is used to fully relax the simulation system. The simulation is subsequently run for 2 × 106Δt to reach a steady heat flow across the simulations system in a constant-volume-and-energy ensemble.
Two-temperature nonequilibrium MD simulation
In this study, the TTM-MD simulation scheme is implemented to perform nonequilibrium MD simulation for phonon degree of freedom, which is coupled with finite difference (FD) calculation for the electron degree of freedom in the LAMMPS package. 40
TTM depicts the coupled electronic and phononic thermal transport through separated temperature fields and a shared coupling term.
13
The temporal and spatial evolution of temperature fields can be described by two coupled heat diffusion equations.
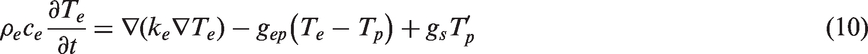
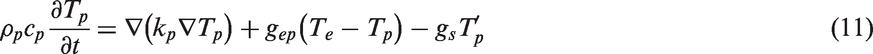
The TTM-MD simulation coupled electronic and phononic subsystems simultaneously as shown in Figure 3. Phononic heat diffusion (equation (11)) is still modeled by the MD technique. As MD simulations only need the inputs of the atomic structure and interatomic potentials, kp with its dependence on temperature and composition is implicitly calculated by MD. The electronic subsystem is modeled by solving equation (10) iteratively with the FD method in the TTM-MD simulations. Phonons and electrons are coupled via the terms Illustration of the coupled electronic and phonic subsystems in TTM-MD simulations. Dots represent atoms in MD simulations, and lines show the FD grid in solving equation (10) using the FD method.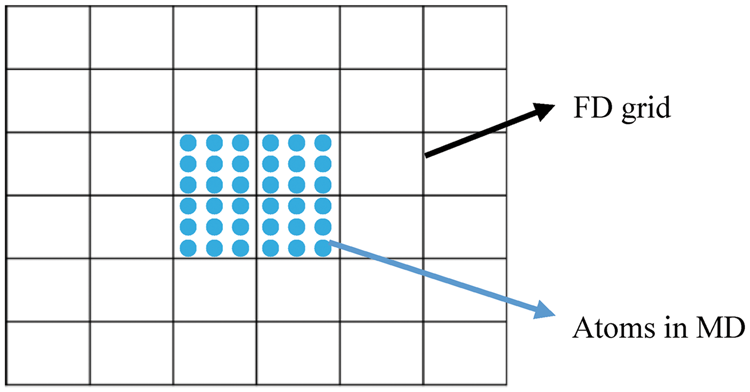
In the implementation of the TTM-MD simulation,
40
the equation of motion for an atom i in the MD simulation is in the form of a Langevin thermostat.
13




In the TTM-MD simulation, the MD part only needs the inputs of atomic structure and interatomic potentials, which can be either measured in the experiments or derived through ab initio methods as discussed in Section “The derivation and selection of interatomic potentials.” On the contrary, the implementation of TTM requires several input parameters as shown in equation (10), i.e. ce, ke,
Multi-scale modeling of thermal conductivity in FE framework
A homogenized 2D FE model is developed to calculate the effective thermal conductivity. The 2D FE model is able to calculate the effective thermal conductivity as a function of temperatures and compositions. The bulk material is represented by continuum elements with embedded thin interfacial elements based on the microstructure characteristics. The thermal conductivity could be evaluated from the FE calculation performed on one RVE as shown in Figure 4. Such a homogenization procedure was used to estimate the effective thermal conductivity of bulk materials, which include large amounts of polycrystals with more details about the FE multi-scale model in the previous study.
44
The SiC particle-reinforced aluminum matrix composites were studied in this paper. More experimental details with respect to SiC particle size and temperature could be referred to the literature.
6
The obtained SiC particle was angular in shape. To facilitate the simulations, spherical SiC particle was assumed as shown in Figure 4. The SiC particle size was represented by the diameter of spherical SiC particle in the multi-scale model. Relatively uniform SiC particle size was found in the experiments for the composites studied here.
6
Our preliminary studies showed that even after considering the uneven distribution of SiC particles in the actual materials,
6
the predicted thermal conductivities still lied within the uncertainties of simulation results shown in this study. Hence, a uniform diameter of SiC particles was modeled in accordance with the observation from the experiments.
6
Multi-scale modeling of SiC-reinforced aluminum matrix composites with varying particle size.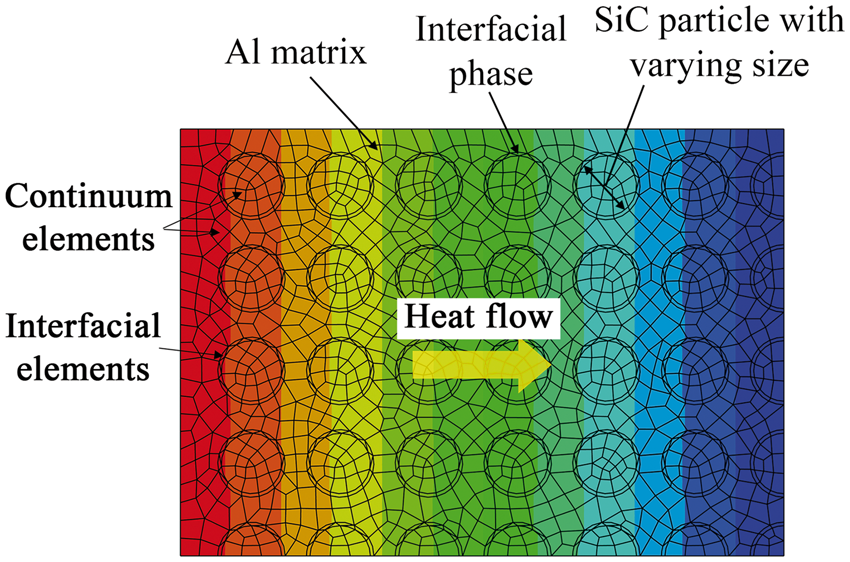
A uniform heat flux was applied at each point of the boundary in Abaqus. For RVE, calculations in the heat flow direction were performed to determine the thermal conductivity, and the thermal conductivity is calculated as
Results and discussion
System size effects in thermal conductivity calculation
One important factor in calculating thermal conductivity is the finite-size effect in MD simulations.14,45 When the length of the simulation cell is not significantly longer than the phonon mean-free path, the actual thermal conductivity will be affected by the system size. 45 This is caused by scattering that occurs at the interfaces with the heat source and sink. To avoid such effect in calculating the interfacial thermal conductivities, studies on the effect of material system size were first performed.
A typical time-average temperature profile used to calculate the thermal conductivity is shown in Figure 5. Within the regions near the heat source or heat sink, nonlinear temperature profiles are observed, which are attributed to the strong scattering caused by the heat source or heat sink.
45
In the intermediate regions, the temperature profiles are fitted with a linear function to obtain the temperature gradients, which are further used to calculate the thermal conductivity by Fourier’s law. The differences in the computed gradients for the different regions are used as an error estimate for the value of thermal conductivity.
Typical temperature profile for 16 × 16 × 2000 Å Al system at room temperature.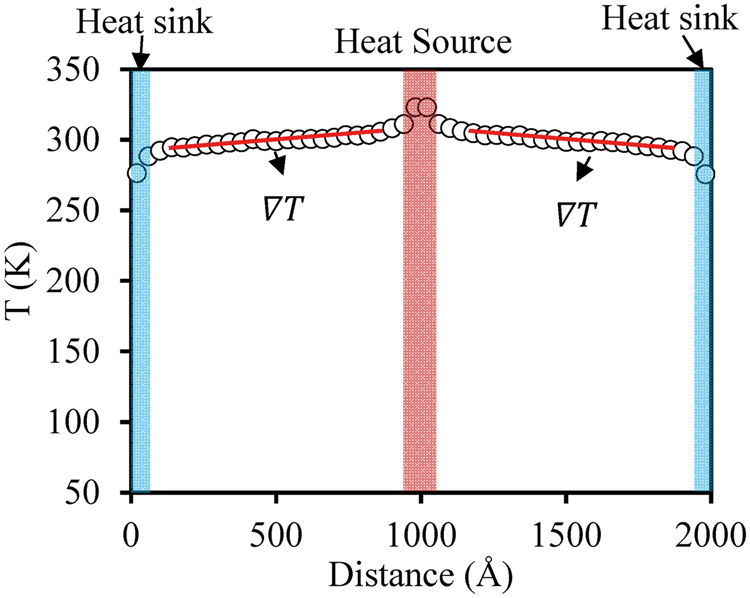
The dependence of k on the dimensions perpendicular to the heat flow, i.e. x/y direction, is first studied. It is expected that k will not depend on the dimensions perpendicular to the heat flow as sensitively as in the direction of the length Lz. Due to periodic boundary conditions on the simulation cell, phonons are free to travel across the simulation cell perpendicular to the heat flow direction without scattering from any boundaries. Hence, changing the dimension perpendicular to the heat flow does not change the scattering in any obvious way. Figure 6 shows a comparison of k obtained for systems of different sizes in the direction perpendicular to the heat flow. The predicted k is almost the same for all the cases. As no obvious dependence on x/y dimensions is observed, the smallest x/y dimensions shown here are used for the rest of studies, i.e. 16 Å.
Comparison of k as for different cell sizes perpendicular to the direction of the heat flow. Each system was 400 Å long parallel to the heat flow in MD simulations. The calculated results at the room temperature for aluminum were shown for demonstration. Similar trend was observed for SiC system.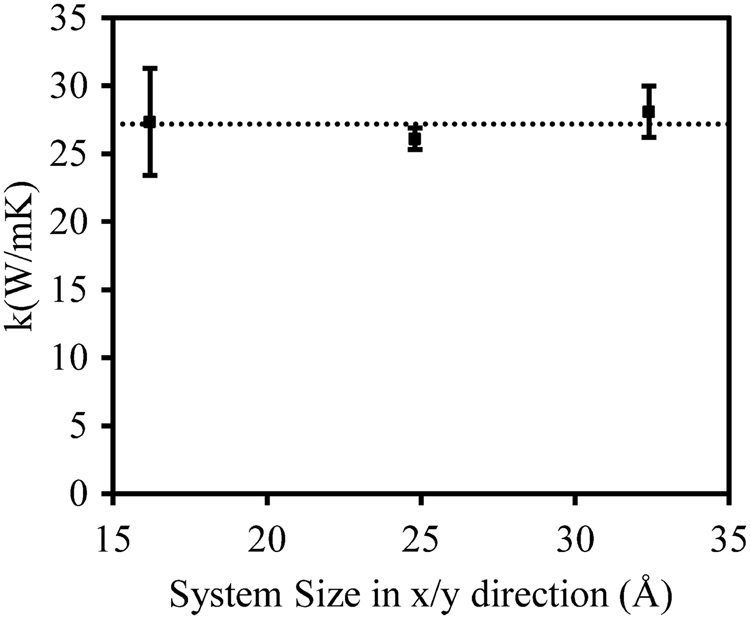
System size dependency of the thermal conductivity of Al.
Calculations of interfacial thermal conductivity across the Al/SiC interface
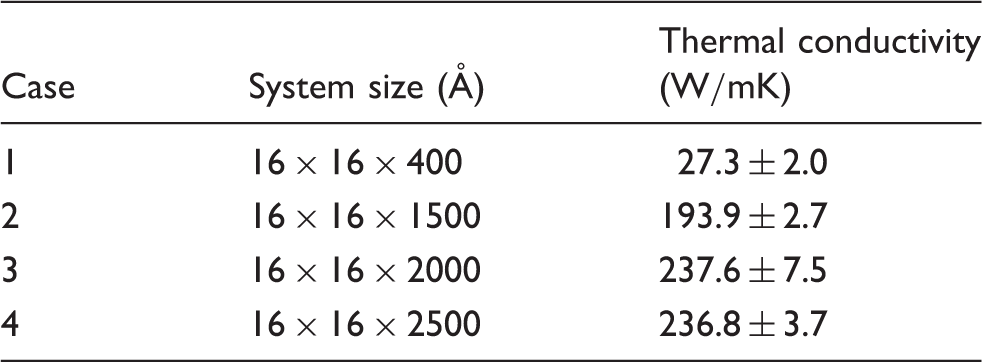
To verify the capabilities of the TTM-MD simulations in modeling coupled electronic and phononic thermal transport across the Al/SiC interface, the thermal conductivity of aluminum was first calculated and compared with experimental measurements. Both conventional MD and TTM-MD simulations were conducted. Thermal conductivity is obtained according to the Fourier’s law of heat conduction. The experimental value and simulation results of k are summarized in Figure 7. It is worth noting that as electrons will not contribute much to thermal transport within SiC and Si, MD simulations without electron effects are sufficient to predict relatively accurate thermal conductivities for SiC and Si.
13
In contrast, the contribution of electrons to thermal transport cannot be ignored for Al, wherein many free electrons exist. k from MD simulations without considering electrons for Al is 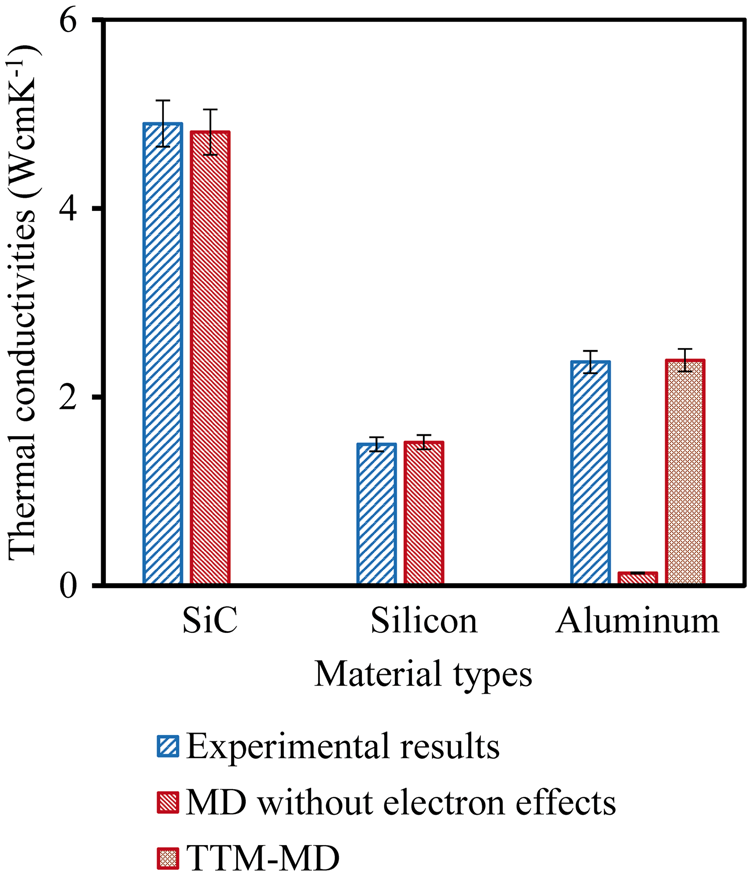
The thermal conductivities of SiC and Si at room temperature were also calculated using the MD simulation method. The MD calculation results were compared with the experimental data in order to validate the proper selection of interatomic potentials. Figure 7 shows the comparison of the predicted thermal conductivities with the experimental data.46,47 It is evident that MD simulations match very well with the experimental measurement with average prediction errors well below 3.0%, thus confirming the validity of interatomic potential selection.
The thermal conductivities of Al/SiC interface at different temperatures were also calculated using the TTM-MD simulation method discussed above. For the NEMD method, it is important to establish a steady-state heat flow. This amounts to obtaining a stationary temperature profile as a function of time, thus ensuring that only steady-state heat flux is flowing. The typical temperature profile of Al/SiC structure is shown in Figure 8. The interface region is defined as the region between Al and SiC based on linear fitting results.
49
The existence of interface contributed to the significant temperature drop between Al and SiC by acting as a thermal barrier for the heat transport between Al and SiC. From the temperature profiles, the temperature drop at the interface, ΔT, can be obtained, which can be subsequently used to calculate the interfacial thermal resistance
A typical temperature profiles of Al/SiC interface structure at room temperature.
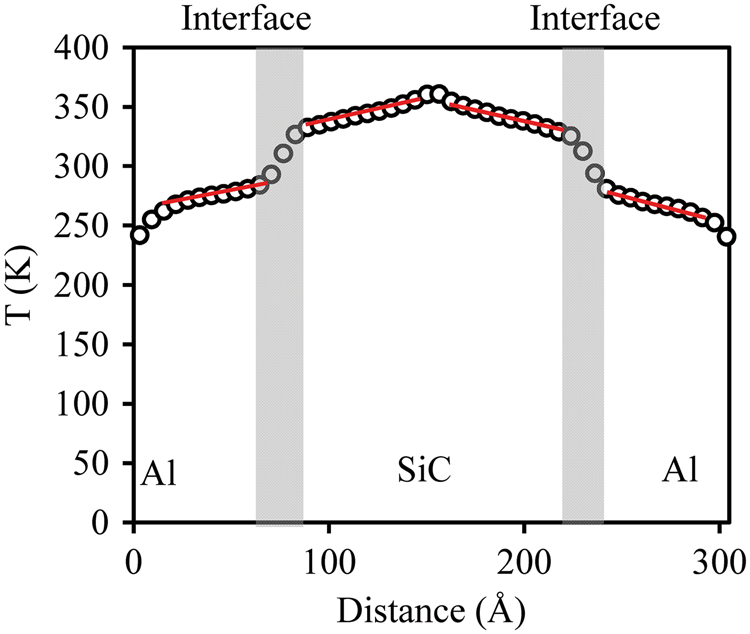
Figure 9 shows a linear relationship between interfacial resistance and inverse system length Lz, as supported by the literature.
14
At the system limit, Al/SiC interfacial resistance as a function of inverse system length at room temperature.
It is worth noting that the interfacial thermal conductivity was also calculated from Fourier’s law as required by the developed multi-scale FE model in this study. The experimental values for Al and SiC are extracted from the measured results from the literature,
6
which are expected to be lower than those of pure Al and SiC materials (as discussed above) due to the existence of impurity in the actual materials. Both the MD results for the interface and the experimental results for Al and SiC are plotted in Figure 10. The interfacial thermal conductivity decreases with increasing temperature as the effect of phonon scattering increases. Due to the increased phonon scattering caused by the interface, the thermal conductivity of interfacial phase is apparently much lower than those of Al and SiC, which is the reason for the much lower thermal conductivity of SiC-reinforced aluminum matrix composites compared with each constituent, i.e. Al matrix and SiC particle, due to the existence of interface between them.
Calculated interfacial thermal conductivities from MD simulations and comparison with experimental measurements of SiC and Al.
6
The inset is an enlargement of the predicted thermal conductivity for Al/SiC interface with respect to temperature.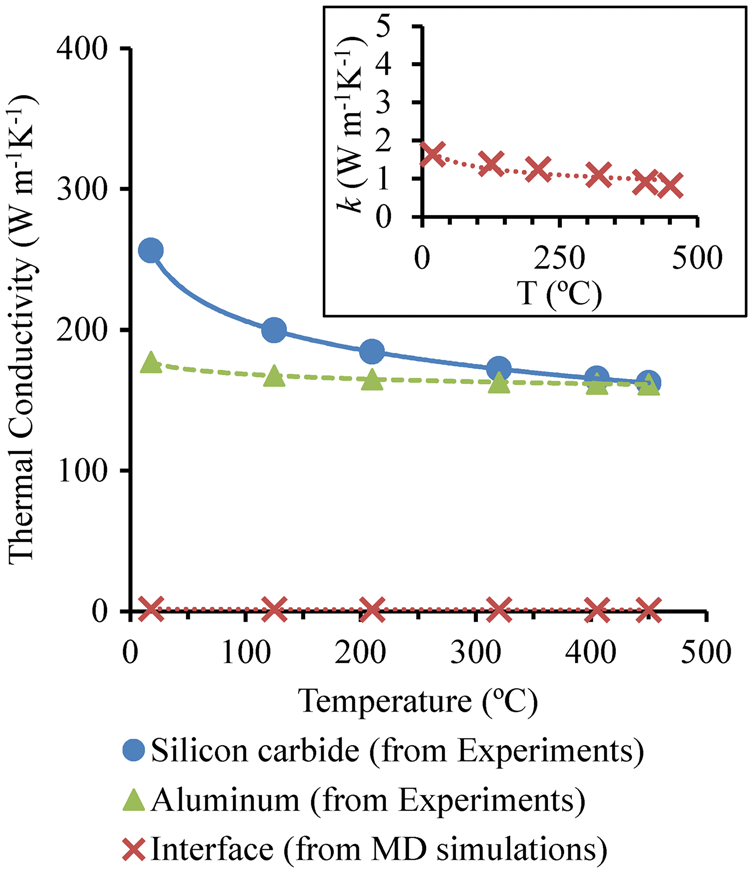
Temperature effects on the thermal conductivities of SiC-reinforced aluminum matrix composites
After interfacial thermal conductivities were obtained, the effective thermal conductivities of SiC particle-reinforced composites at different temperatures were calculated using the developed multi-scale model in Section “Multi-scale modeling of thermal conductivity in FE framework.” The temperature-dependent thermal conductivities for both SiC particles and Al matrix
6
were also considered in the simulations. The calculated results are shown in Figure 11, where it can be seen that the simulated results of the multi-scale model yield a maximum 6.21% deviation compared to experimental results. The predicted results are consistently higher than the experimental measurements, which can be explained by the defects and voids existing in the actual composites. Since only ideal composite microstructure was considered in the simulations, the existence of defects and voids can lead to phonon scattering, lowering the thermal conductivity of the actual material.
Comparison between effective thermal conductivities and experimental measurements
6
for 40 vol% particulate-SiC-reinforced aluminum matrix composites. The SiC particle size is 10.2 µm in diameter.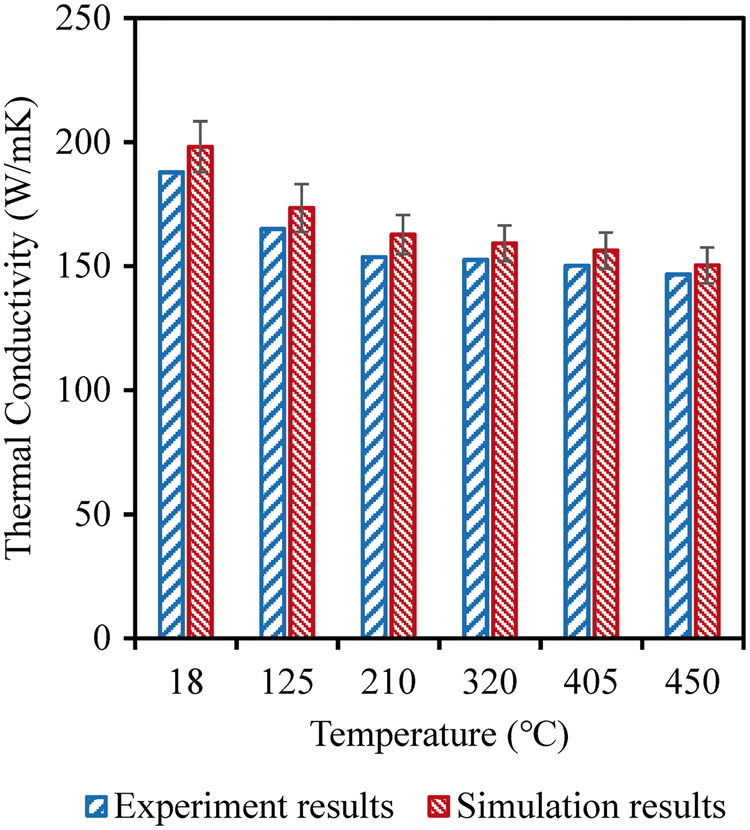
Effect of SiC reinforcement particle size on thermal conductivities of aluminum matrix composites
The effects of SiC particle size on the effective thermal conductivity were studied at room temperature. As shown in Figure 12, calculations of effective thermal conductivities show an increase in effective thermal conductivities with increasing SiC particle size as found experimentally by Hasselman et al.
6
While particle sizes increased, the SiC volume fraction remained the same as 40 vol% in the composite. The prediction results show a good agreement with the experimental measurements as shown in Figure 12.
Comparison between effective thermal conductivities and experimental measurements
6
for 40 vol% SiC-reinforced aluminum matrix composites with different SiC particle size at room temperature.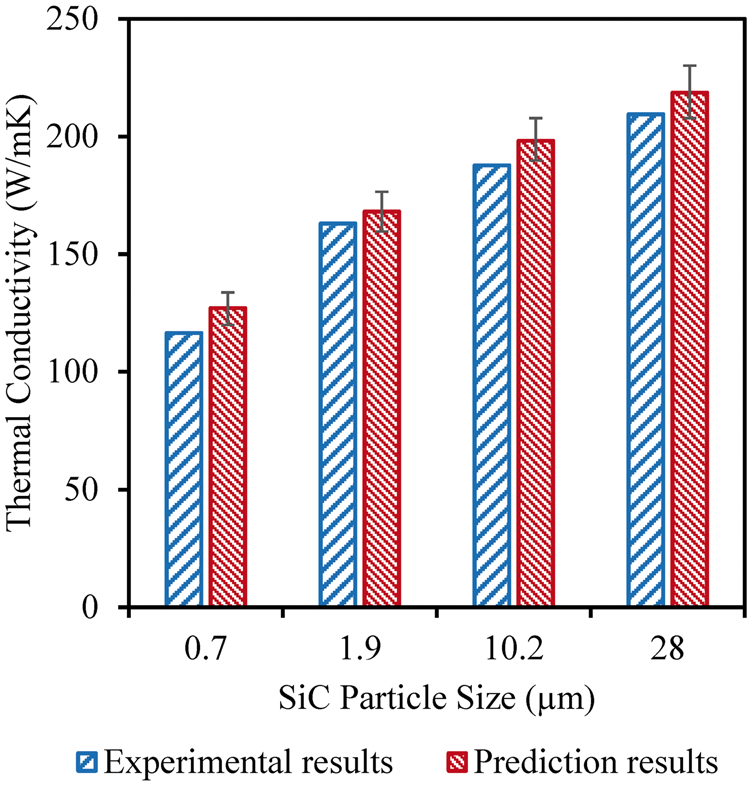
The increasing thermal conductivity with increasing particle size can be explained by the fact that with increasing SiC particle size and associated decrease of total interfacial area, the relative contribution of interfacial thermal barrier to the total conductivity of the composite decreases, thus yielding an increasing effective thermal conductivity. The maximum discrepancy between experimental measurements and prediction results is within 9.88%, thus validating the capability of the multi-scale modeling in predicting the effective thermal conductivity.
Effect of SiC volume fraction on thermal conductivities of aluminum matrix composites
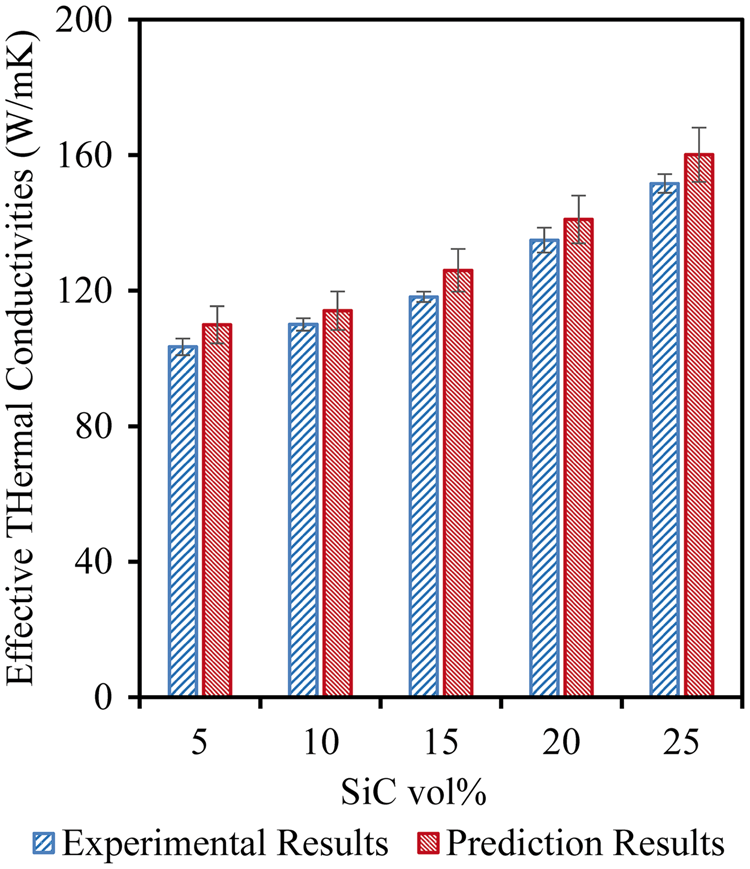
Studies on the effect of SiC volume fraction were also carried out using the developed multi-scale model. A nominal particle size of 37 µm
54
was implemented in modeling SiC-reinforced aluminum matrix composites. SiC particles were uniformly distributed in the aluminum metal matrix composite with varying SiC volume fractions from 5% to 25%. The variations of effective thermal conductivities are plotted in Figure 13. It can be seen in Figure 10 that the thermal conductivity of SiC is relatively larger than that of Al. As the SiC volume fraction increases, its relative contribution to the total conductivity of the composites increases, hence enhancing the effective thermal conductivity of the composites. As seen in Figure 13, the prediction results agreed well with experimental measurements with differences being smaller than 7.48%.
Effect of SiC volume fraction on effective thermal conductivities and comparison with experimental results.
54
Conclusion
A multi-scale model has been developed to predict the thermal conductivity of SiC-reinforced aluminum metal matrix composites. An ab initio method was used to calculate the interatomic potential for MD simulations. The interfacial thermal conductivity was calculated in the TTM-MD simulations, which was subsequently used in the proposed multi-scale model to predict the thermal conductivity of bulk materials. The model successfully predicted the decreasing thermal conductivity with increasing temperature. A small effect of electronic contribution to the thermal transport of the Al/SiC interface was observed. It has also been shown that the increase of SiC particle size and volume fraction can increase the thermal conductivity due to the reduced interfacial thermal barrier and the increased contribution of SiC reinforcement, respectively. The prediction results agreed well with the measurements for different cases, showing a maximum of 9.88% discrepancy. The successful predictions of thermal conductivity proved the efficacy of the developed multi-scale model, which can be used to design thermal properties of ceramics and composites with different compositions.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.