
Abstract
The odontogenic keratocyst (OKC, currently designated by the World Health Organization as a keratocystic odontogenic tumor) is a locally aggressive, cystic jaw lesion with a putative high growth potential and a propensity for recurrence. Although it is generally agreed that some features of OKCs are those of a neoplasia, notably the relatively high proliferative rate of epithelial cells, controversies over the behavior and management of OKCs still exist. This article is intended to review this intriguing entity and to summarize the findings of recent studies related to the nature of OKCs and their clinical and therapeutic implications. Recent advances in genetic and molecular research, i.e., PTCH1 mutations and involvement of the Hedgehog signaling pathway, have led to increased knowledge of OKC pathogenesis which hints at potential new treatment options, although the question of whether the OKC is a cyst or a cystic neoplasm is yet to be answered with certainty. Since some advocate a more conservative treatment for OKCs, notably marsupialization and decompression, future treatment strategies may focus on molecular approaches and eventually reduce or eliminate the need for aggressive surgeries.
Keywords
Introduction
The term ‘odontogenic keratocyst’ (OKC) was first introduced by Philipsen over 50 years ago to describe a group of odontogenic cysts which showed a characteristic histological appearance (Philipsen, 1956). As compared with other types of odontogenic cysts, OKCs appear to have an intrinsically higher growth potential (Main, 1970; Browne, 1971; Li et al., 1993, 1994a,b). A propensity to recur following surgical treatment, a relationship to the so-called ‘Gorlin syndrome’ (also known as nevoid basal cell carcinoma syndrome), and the potential risk of neoplastic change place OKCs in a unique position within the spectrum of odontogenic lesions. It has long been suggested that OKCs should be regarded as benign neoplasms (Ahlfors et al., 1984; Shear, 2002a,b). While the first 2 WHO classifications of odontogenic lesions (Pindborg et al., 1971; Kramer et al., 1992) put OKCs into the category of developmental odontogenic cysts, the most recent edition designates the OKC as a keratocystic odontogenic tumor (Barnes et al., 2005), implying that the lesion is a benign neoplasm. This new classification and terminology have aroused heated debates over the nature of OKCs, particularly among oral and maxillofacial surgeons and pathologists. Although considerable insight into the biological profile of the OKC has been accumulated in recent years (Gomes et al., 2009a; Mendes et al., 2010), controversies over its behavior and management still exist. The question of whether the OKC is a cyst or a cystic neoplasm is yet to be answered with certainty. This article was developed to provide an overview of the paradoxical aspects of OKCs and to review the recent advances related to the discussions over the nature of OKCs and their clinical and/or therapeutic implications.
Clinico-Pathological Profile
Overall, OKCs probably account for 8-11% of odontogenic cysts. They are present in patients over a wide age range, but peak in the 2nd and 3rd decades and are more common in males. The mandible is the most common site (Browne, 1971; Luo and Li, 2009). About half of all OKCs occur at the angle and ramus of the mandible. In the maxilla, most OKCs present in the so-called ‘globulomaxillary’ and molar areas. A considerable number of OKCs are asymptomatic and hence are detected only by incidental radiographic findings. When they are symptomatic, swelling and intra-oral drainage appear to be most common. The radiographic presentation of OKCs is variable. Typical features, such as scalloped margins or a multilocular appearance (Fig. 1A), are indicative, but other odontogenic lesions may show similar radiological findings. Thus, there are few unequivocal clinical and radiographic features specific for OKCs. Definitive diagnosis still relies on histological examination.
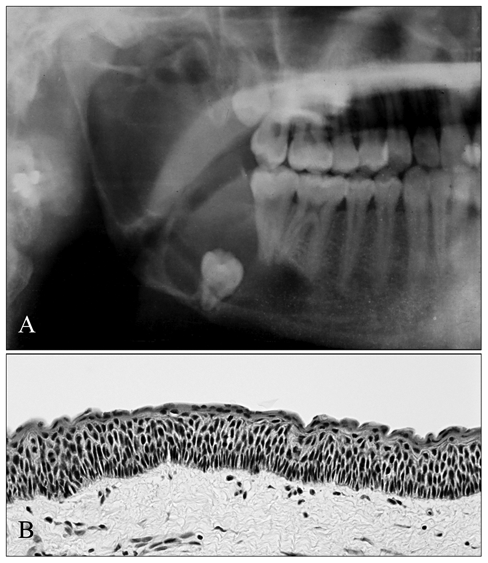
Radiograph of a mandibular OKC, involving the angle and ramus region, depicts a multilocular radiolucency associated with an impacted third molar
Typical histologic features of OKCs have been well-characterized (Philipsen, 1956; Browne, 1971), including: a thin, uniform lining of stratified squamous epithelium with a tendency to detach from the underlying connective tissue capsule; a thin corrugated surface layer of parakeratin; a spinous cell layer 8 to 4 cells in thickness, often showing intracellular edema; a regular layer of columnar basal cells with nuclear palisading; a flat epithelial-fibrous tissue junction, usually devoid of epithelial rete ridges; and a relatively thin fibrous capsule that mostly lacks inflammatory cell infiltrate (Fig. 1B). The OKC is of particular interest because of its clinically more aggressive behavior and tendency to recur after surgery. The incidence of recurrence in various reported series has varied from 2.5 to 62% (Browne, 1991; Shear, 2002a). The reason for this great variation is partly due to the varied nature of the cases published. For example, some series included cysts from patients with Gorlin syndrome, and others excluded them. Other important variables include the duration of the follow-up periods and the methods of treatment used.
Although an OKC most commonly occurs as a single lesion in the jaw of an otherwise healthy person, about 4-5% of all OKC patients have multiple cysts with other features of the so-called ‘Gorlin syndrome’ (Gorlin and Goltz, 1960; Browne, 1991). Gorlin syndrome is a rare autosomal-dominant disorder that exhibits high penetrance and variable expressivity. Clinical manifestations are extremely varied and include basal cell carcinoma of the skin, multiple OKCs of the jaws, palmar or plantar pits, and ectopic calcification of the falx cerebri, which are considered major criteria for diagnosis (Gorlin, 1995). Multiple OKCs are the most consistent and common anomaly in Gorlin syndrome, occurring in 65-100% of patients, often during the first or second decade of life (Woolgar et al., 1987). In addition, syndrome-associated OKCs are to be found in both jaws with equal frequency, in contrast to sporadic OKCs, which involve especially the lower jaw (Lo Muzio et al., 1999a). Among the various presentations, OKCs are often the first signs of Gorlin syndrome, frequently antedating the syndromic basal cell carcinomas, thereby allowing for earlier diagnosis (Lo Muzio et al., 1999b).
Evidence accumulated from various clinico-pathological observations suggests that the OKC should be regarded as a neoplasm, due to its local aggressiveness, a high tendency to recur, its association with Gorlin syndrome, and the occasional occurrence of malignant transformation (Ahlfors et al., 1984; Dabbs et al., 1994; Makowski et al., 2001; Shear, 2002a,b).
Epithelial Cell Proliferation and Differentiation
An important piece of evidence supporting the neoplastic nature of OKCs is the consistent detection of a higher level of cell-proliferative activity in the lining epithelium. Early histological studies had shown that mitotic figures are a prominent feature of OKC epithelium (Main, 1970; Browne, 1971). However, mitosis represents only the shortest phase of the cell cycle; thus its index may not be sensitive enough to reflect cellular activity. Further evidence of a greater epithelial activity in OKC linings was produced by Toller (1971), who estimated tritiated thymidine uptake in explants of cyst walls by autoradiography. The mean labeling index, expressed as the mean percentage of labeled cells per 1000 basal cells, was 13.0%, which was approximately 7 times greater than that for non-keratinizing jaw cysts (1.7%).
Immunocytochemical methods of assessing cell proliferation have particular advantages over other techniques because of the maintenance of cellular and tissue architecture, the relative simplicity of methodology, and the rapidity of results. Using quantitative techniques based on a combination of digital image analysis for histomorphometric measurement of basement membrane and manual counting of PCNA and Ki67 immunostained cells, Li et al. investigated the proliferative activity in the epithelial linings of various major and/or sub-types of odontogenic jaw cysts (Li et al., 1994a,b, 1995). The results demonstrated that OKC linings exhibited a significantly higher level of PCNA labeling, with a predominantly suprabasal location of positive cells in comparison with dentigerous and radicular cyst linings (Figs. 2A, 2B). A comparison of different subtypes of OKC linings indicated an increased number of cells expressing Ki67 in Gorlin-syndrome-related OKC than in those of non-syndromic OKCs. Interestingly, however, no significant difference was found between recurrent and non-recurrent lesions of non-syndromic OKCs (Li et al., 1995) (Fig. 2C). The demonstration of a similar Ki67 labeling index in the linings of recurrent and non-recurrent OKCs indicates that recurrence is not associated with a subgroup of lesions showing increased proliferation, supporting the concept that inappropriate surgery on the original cyst is the most plausible reason for recurrence (Li et al., 1995). Using a semi-automated image analysis system, Landini (2006) compared the epithelial lining architecture of OKCs with that of radicular cysts. While a significant difference in the quantitatively estimated thickness in the number of cell layers between linings of OKCs and radicular cysts was detected, there was no significant difference between Gorlin-syndrome-related OKCs and sporadic OKCs. Thus, the increased proliferation does not appear to be related to the thickness of syndrome-related OKC linings, which may suggest a rapid epithelial turnover. The heightened proliferative activity of Gorlin-syndrome-related OKCs could reflect the underlying genetic abnormalities in this group of individuals. (See later section regarding PTCH1.)
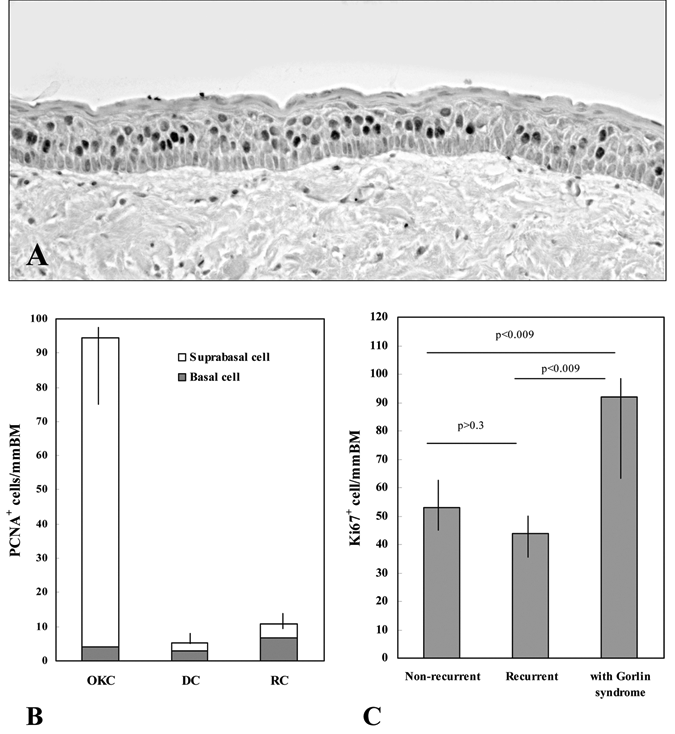
Immunocytochemical staining of PCNA in OKC lining epithelium (
The consistently higher level of PCNA and Ki67 labeling in the epithelial linings of OKCs supports the hypothesis that active cell division of the lining epithelium or mural growth is more important in the pathogenesis of OKCs than in other types of odontogenic cysts. The characteristic suprabasal location of the proliferating cells predominant in OKC linings, in contrast to that in dentigerous and radicular cysts, suggests that a unique cellular proliferation and/or differentiation process occurs within this cyst type. Histologically, the columnar structure and the apparent reverse polarity of basal cells of OKC epithelium resemble the pre-ameloblasts in developing enamel organ, suggesting that they may, to some extent, undergo ameloblastic differentiation. Studies involving the transplantation of the OKC tissue walls into athymic mice have demonstrated that the characteristic features of the epithelium are retained only in the presence of its own fibrous tissue capsule (Vedtofte et al., 1982). It is therefore interesting to suggest that the unique, predominantly suprabasal distribution of proliferating cells within OKC epithelia may be a consequence of ameloblastic differentiation within the basal cell layer, due to inductive influences of underlying connective tissue (Li et al., 1994b, 1995). The nature and possible role of these inductive influences in epithelial cell proliferation and differentiation within OKCs remain to be determined.
The p53 Gene
The p53 gene product is thought to control cell growth, with its wild-type form arresting cell cycles at the G1 phase and its mutant forms promoting cell proliferation and/or malignant transformation (Lane and Benchimol, 1990; Levine et al., 1991). It is therefore interesting to determine whether abnormalities of the p53 gene are associated with the development of OKCs. Possible involvement of the p53 gene in the growth and regulation of OKCs has been suggested by immunocytochemical demonstration of p53 protein overexpression in OKC lining cells (Ogden et al., 1992; Li et al., 1994a). Quantitative study of p53 immunoreactivity demonstrated a significantly higher level of p53 labeling in OKC linings as compared with that in dentigerous and radicular cysts (Li et al., 1996), which significantly correlated with Ki67 labeling within the same series of cyst cases. Unless molecular analysis of the p53 gene is performed, however, it is unknown whether the increased p53 labeling in OKC epithelium indicates mutation of the p53 gene or overexpression of the wild-type product due to stabilization by other gene products, e.g., cdc2 protein kinase (Sturzbecher et al., 1990) or the mdm2 gene product (Wu et al., 1993). Thus, Li et al. (1996) further examined the status of the p53 gene in the immunopositive OKC cases using combined polymerase chain-reaction and single-stranded conformation polymorphism (PCR-SSCP), followed by DNA direct sequencing. The results indicated that DNA extracted from OKC, including Gorlin-syndrome-associated lesions, harbored no mutations in the p53 gene.
In other words, the epithelial lining of the OKC expresses higher levels of p53 protein than do other cyst types, which appears to correlate with cell proliferation. However, this overexpression is not due to mutation of the p53 gene, but presumably reflects overproduction and/or stabilization of normal p53 protein. As hypothesized by some authors, a high proliferation rate may result in detectable concentrations of wild-type p53 protein in cells (Mercer and Baserga, 1985; Villuendas et al., 1992). This is supported by the experimental finding of detectable levels of wild-type p53 protein in phytohemagglutinin-stimulated, rapidly proliferating lymphocytes (Mercer and Baserga, 1985) and by the occurrence of sporadic p53-positive cells in thymus and reactive lymphoid tissues (Villuendas et al., 1992). It is, therefore, reasonable to believe that the overexpression of p53 by OKC epithelium may represent a ‘feedback’ response to its high proliferative activity, rather than the cause of its intrinsic growth potential.
The PTCH1 Gene Mutation and Involvement of the Hedgehog Signaling Pathway
Perhaps the most important and intriguing evidence that appears to tip the balance of the argument over the nature of OKCs is the investigation of the PTCH1 gene. This gene is the human homologue of the Drosophila segment polarity gene, patched, which has been proven to be the disease-causing gene for the Gorlin syndrome (Hahn et al., 1996; Johnson et al., 1996). PTCH1 has been mapped to 9q22.3–31, consisting of 23 exons and encoding a transmembrane protein of 1447 amino acids with 12 transmembrane regions, 2 extracellular loops, and a putative sterol-sensing domain (Hahn et al., 1996; Johnson et al., 1996). Like neoplasms in other cancer predisposition syndromes, OKCs in Gorlin syndrome patients are multiple and appear in a random pattern; similar isolated defects are also seen occasionally in the general population. Our studies (Gu et al., 2006; Li et al., 2008; Sun et al., 2008; Pan and Li, 2009), as well as those of others (Barreto et al., 2000; Ohki et al., 2004; Song et al., 2006), have revealed that over 85% of syndromic OKCs and nearly 30% of sporadic OKCs harbored PTCH1 mutations (Table). These findings are in general agreement with those of previous studies in patients with syndromic and/or sporadic basal cell carcinomas (Lindström et al., 2006). Most of the identified frameshift or nonsense mutations lead to the synthesis of a truncated PTCH1 protein. These mutations appear to be mainly clustered into the 2 large extracellular loops of the PTCH1 protein (Fig. 3), which are important functional domains to bind Sonic hedgehog ligand. But no apparent genotype-phenotype correlation has been established (Lindström et al., 2006; Pan and Li, 2009). In addition, loss of heterozygosity (LOH) at chromosome 9q22–31, the region to which the PTCH1 gene maps, has been observed as a frequent event in syndrome-associated tumors, including OKCs (Chenevix-Trench et al., 1993; Levanat et al., 1996; Pan et al., 2010). These findings indicate that defects of PTCH1 are involved in the pathogenesis of syndromic as well as sporadic OKCs.
Summary of PTCH1 Mutations in Gorlin-syndrome-related and Sporadic OKCs (GenBank U59464.1) a (numbering of nucleotides: +1 = A of ATG codon)
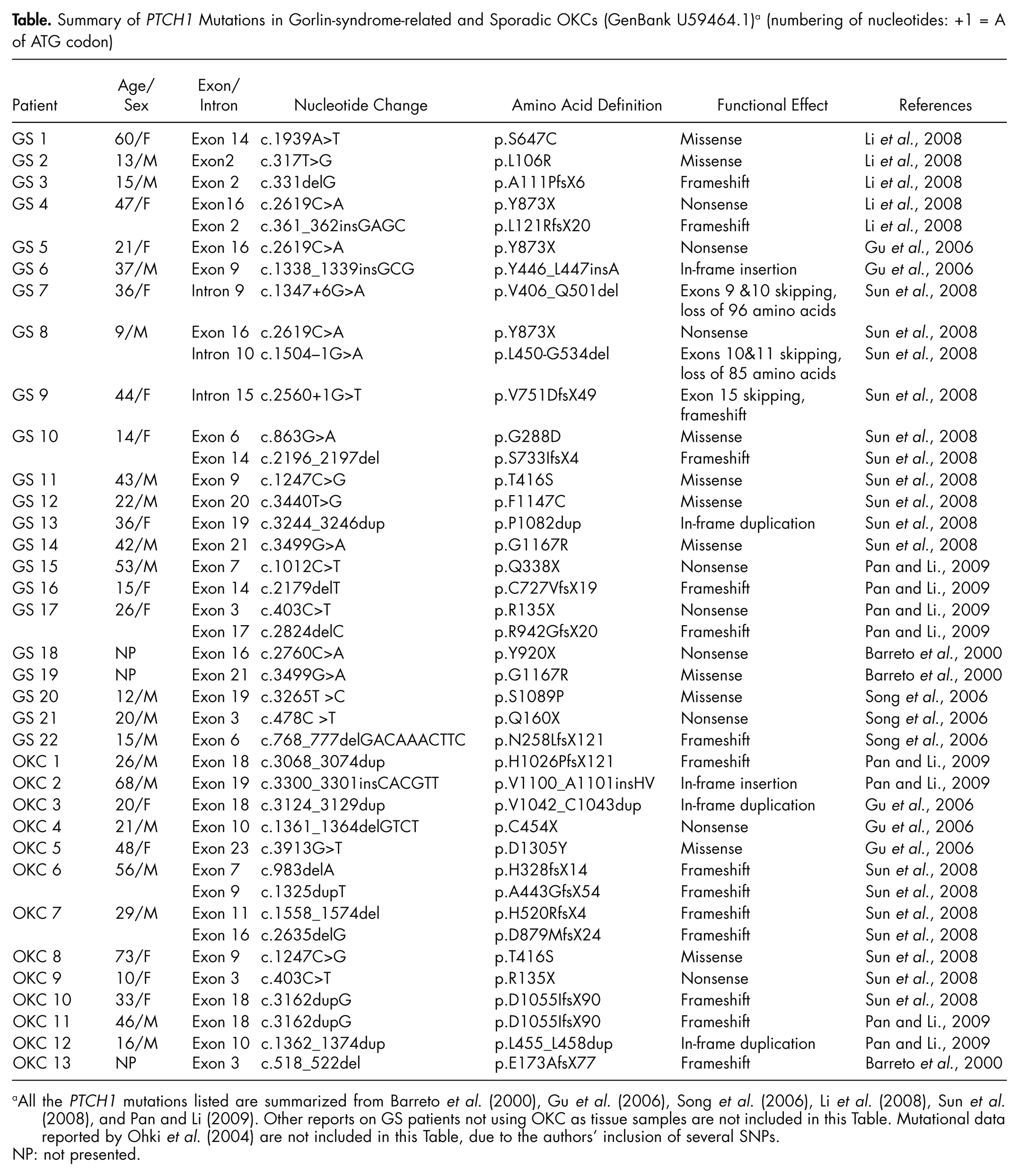
All the PTCH1 mutations listed are summarized from Barreto et al. (2000), Gu et al. (2006), Song et al. (2006), Li et al. (2008), Sun et al. (2008), and Pan and Li (2009). Other reports on GS patients not using OKC as tissue samples are not included in this Table. Mutational data reported by Ohki et al. (2004) are not included in this Table, due to the authors’ inclusion of several SNPs.
NP: not presented.
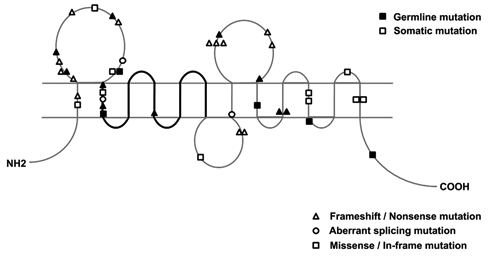
Distribution of PTCH1 mutations (germline, black; somatic, blank) in GS-related and sporadic OKCs, reported by our group and others listed in the Table, in relation to the domain structure of the PTCH1 protein. The triangle denotes a nonsense or a frameshift mutation, the circle denotes a splice mutation at the adjacent exon junction, the square denotes a missense mutation or in-frame insertion/deletion, and the thick line denotes the sterol-sensing domain.
The aggressive behavior and higher tendency for recurrence of OKCs have been attributed to the greater proliferative activity of the epithelial linings. To clarify the role of PTCH1 mutations in OKCs, investigators have studied epithelial cell proliferation as assessed by Ki67 labeling in a total cohort of 62 OKCs (42 sporadic and 20 syndromic cases) with known PTCH1 status (Pan and Li, 2009). The epithelial Ki67 labeling in OKCs with PTCH1 mutation was significantly higher than that in cases with no PTCH1 mutation. Furthermore, OKCs harboring PTCH1 truncation-causing mutations showed an even greater Ki67 labeling index than those with non-truncation-causing mutations. These results suggest that PTCH1 mutations, particularly those causing protein truncation, are associated with OKCs showing increased proliferative activity, and thus may relate to a phenotype of higher recurrent tendency (Pan and Li, 2009).
PTCH1 is an important molecule in the so-called Hedgehog (Hh) signaling pathway. This pathway is a key regulator of embryonic development controlling cell proliferation and cell fate. As a receptor for the Sonic hedgehog protein (SHH), PTCH1 inhibits the signaling pathway by repressing the activity of Smoothened (SMO), a seven-span transmembrane protein with homology to a G-protein-coupled receptor (Stone et al., 1996) (Fig. 4A). According to this model, loss of PTCH1 function by inactivating PTCH1 mutations as well as aberrant activation of SMO by activating SMO mutations could cause constitutive, ligand-independent signal transduction that may lead to neoplastic growth (Toftgård, 2000) (Fig. 4B). In contrast to the frequent detection of PTCH1 mutations in OKCs, no pathogenic SMO mutation was detected in a total of 50 OKCs (Sun et al., 2008), among which were 16 Gorlin syndrome patients. These results suggest that mutation in SMO is extremely rare and that misregulation of the Hh signaling caused by PTCH1 inactivation is involved in the pathogenesis of OKCs (Sun et al., 2008; Pan et al., 2010).
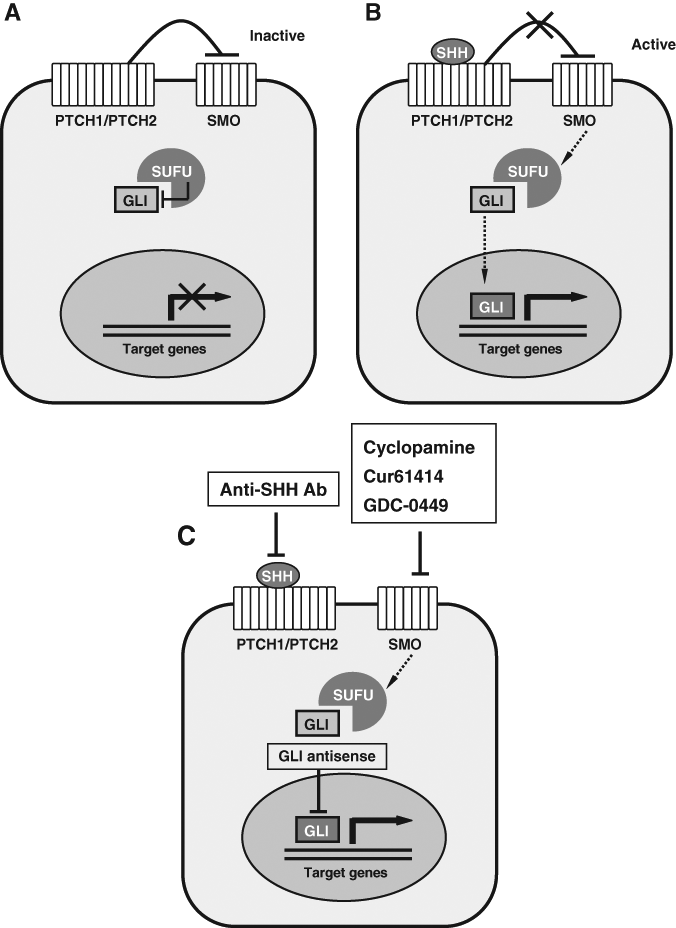
Hedgehog (Hh) signaling pathway. In the absence of ligand, the Hh signaling pathway is inactive
According to Knudson’s two-hit model of tumor suppressor genes (Knudson, 1971), two mutations—one occurring in each of the 2 alleles of the gene, or one mutation in one allele accompanied by another allelic loss of the remaining wide-type allele—are required to trigger neoplasm formation. The molecular analysis of PTCH1 in Gorlin-syndrome-associated tumors suggests that a ‘two-hit’ hypothesis is applicable to their pathogenesis (Levanat et al., 1996). These syndrome-associated tumors presumably arise from precursor cells that contain a hereditary ‘first hit’. Additional somatic mutation, LOH, or epigenetic silence of the other allele, acting as the ‘second hit’, would introduce functional inactivity of the PTCH1 protein. As a test of this hypothesis, a range of PTCH1 alteration profiles, including genetic mutation, LOH, and promoter hypermethylation, was recently investigated by Pan et al. (2010) to elucidate the possible genetic and epigenetic mechanisms of PTCH1 inactivation in syndromic and non-syndromic OKCs. Of all the 44 OKC samples (15 syndromic and 29 sporadic cases) tested, 13 cases (30%) were identified to fit the standard two-hit model, 10 of which contained 2 inactivating alleles by LOH and mutation separately, and 3 obtained disrupted alleles through 2 mutations in each of the alleles. Fourteen OKCs (32%) were found to conform to a one-hit model by LOH or mutation in a single allele, 5 of which lost a normal allele in PTCH1 locus, 7 tumors carried only one mutation, and 2 tumors harbored 2 coincident mutations in the same allele of PTCH1. After careful analysis, the remaining 17 cases (38%) failed to show any PTCH1 alteration. In addition, hypermethylation of the PTCH1 promoter was not identified in their series (Pan et al., 2010). These results suggest that numerous mechanisms are implicated in inactivation of the PTCH1 gene, in addition to the two-hit hypothesis. Perhaps the most contested exception to the two-hit hypothesis is haplo-insufficiency (absent or reduced function due to the loss or inactivation of a single allele). Evidence for the haplo-insufficiency model arises from studies of PTCH1 +/− mice (Wetmore et al., 2000; Zurawel et al., 2000). Mice heterozygous for PTCH1 recapitulate the typical developmental symptoms of Gorlin syndrome and develop medulloblastoma, implying that haplo-insufficiency of PTCH1 is sufficient to promote tumor formation in mice. In vivo studies have demonstrated that several mutant PTCH1 proteins could result in activation of Hh signaling through a dominant-negative mechanism, despite the production of wild-type PTCH1 (Hime et al., 2004). Thus, not only the standard two-hit model, but also the one-hit model exhibiting haplo-insufficiency or dominant-negative isoforms might be involved in the inactivation of the PTCH1 gene. The failure to detect any hit in about one-third of OKC cases examined might be explained by the presence of a multi-gene tumorigenesis model.
It is believed that tumors arise from a multi-step progression. The more advanced the tumor, the more hits it had accumulated. Our group has recently demonstrated that the percentage of two-hit cases in syndromic OKCs (53.3%) was significantly higher than that in sporadic OKCs (17.2%) (Pan et al., 2010), suggesting that more destructive hits accumulated due to decreased DNA repair efficiency and/or increased genetic instability in syndrome-related lesions. The cases with only one hit at the time might represent an early stage of tumor progression, and a second hit might eventually occur with a more severe phenotype. Thus, the evidence is mounting that genetic alterations of PTCH1 and the Hh pathway are involved in the development of OKCs. All these changes, i.e., PTCH1 mutations, LOH, two-hit mechanisms, and so on, are by definition features of a neoplastic process. However, one may still argue that these detected abnormalities are not consistent in all OKCs, but are seen in only a proportion of cases. Furthermore, many known non-neoplastic entities could harbor specific genetic abnormalities, such as SH3BP2 gene mutations in cherubism (Ueki et al., 2001) and GNAS1 gene mutations in fibrous dysplasia (Riminucci et al., 1999). All these raise the question of whether neoplasia can be defined by molecular genetic criteria.
Genetic Alterations in Other Genes
Although PTCH1 has been identified to play a confirmative role in Gorlin syndrome, other genes, such as PTCH2 (Fan et al., 2008; Xu and Li, 2008) and SUFU (Pastorino et al., 2009), might also be involved. PTCH2 encodes a putative transmembrane protein of 1203 amino acids, which has high homology to the PTCH1 protein (Smyth et al., 1999). Similar to PTCH1, PTCH2 is a receptor for SHH, and is involved in SHH/PTCH cell signaling (Carpenter et al., 1998) (Fig. 4A). Fan et al. (2008) identified a novel PTCH2 mutation in a Chinese Gorlin syndrome family, resulting in the loss of PTCH2 inhibitory function in the Hh signaling pathway. Our group also identified 2 novel PTCH2 missense mutations in 15 unrelated Gorlin syndrome patients. Interestingly, one PTCH2 mutation occurred in a patient carrying no PTCH1 mutation, but the other case carried both PTCH1 and PTCH2 mutations (Xu and Li, 2008). SUFU, encoding the human ortholog of Drosophila suppressor of fused, is a negative regulator of Hh signaling that interacts with all 3 GLI proteins (Fig. 4A) and mediates their nuclear export in the absence of SHH (Dunaeva et al., 2003). SUFU is a tumor-suppressor gene that predisposes individuals to medulloblastoma (Taylor et al., 2002). Pastorino et al. (2009) identified a SUFU germline splicing mutation in a family that was PTCH1-negative and who had signs and symptoms of Gorlin syndrome. Thus, although not as frequent as PTCH1 mutation, PTCH2 and SUFU germline mutations are detectable in a small number of Gorlin syndrome families. Whether these alterations represent clinically distinct phenotypes of Gorlin syndrome, in comparison with PTCH1 mutations, remains to be clarified with identification of additional families. Genotypic analysis performed by different research groups also demonstrated loss of heterozygosity in p16, p53, MCC, TSLC1, LTAS2, and FHIT genes (Agaram et al., 2004; Henley et al., 2005; Malcic et al., 2008). All of these genes are tumor-suppressor genes associated with different types of human neoplasia. These findings are helpful to explain the aggressive behavior of the OKC and give further support for its neoplastic nature.
Clonality Analysis
A distinguishing feature of a neoplasm is its origin from a single clone of genetically identical cells (Nowell, 1976; Secker-Walker, 1985). Thus, defining clonality provides insight into the mechanisms of cellular proliferation and growth characteristics of “proliferative” lesions. Clonality can be inferred by referencing clonal markers such as the pattern of X chromosome inactivation in lesional tissue from female patients (Allen et al., 1992). The X-linked human androgen receptor (HUMARA) gene contains a polymorphic DNA marker that reliably illustrates the pattern of X chromosome inactivation in a tissue. Normal tissues are composed of cell populations that contain random X inactivation patterns of both paternal and maternal X-chromosomes, whereas a monoclonal cell population of a neoplastic lesion contained a non-random inactivation pattern, derived either from the mother or from the father. Using this technique, Gomes et al. (2009b) investigated the clonal origin of 19 cases of odontogenic tumors, including 6 cases of OKC. Among the 16 informative cases, 12 showed a monoclonal pattern. Four of the six OKCs were monoclonal, and the remaining two were polyclonal. While emphasizing that most odontogenic tumors are clonal, including OKCs, the authors did attribute the four polyclonal cases to the contamination of samples by stromal or inflammatory cells. The two polyclonal OKCs contained a significant dispersed inflammatory cell component (Gomes et al., 2009b). Current work undertaken in the author’s laboratory also produced a rather mixed picture regarding the clonal pattern of OKCs, although the majority of OKCs appeared to be monoclonal (unpublished observations). These findings do indicate that considerable numbers of OKCs are composed of a monoclonal population of cells, lending support for their neoplastic nature.
New Treatment Strategies
The treatment of OKCs remains controversial. The challenges lie in minimizing both the risk of recurrence and the surgical morbidity. Numerous modalities, ranging from decompression alone, to simple enucleation with or without curettage, to resection, have been used in the management of OKCs (Morgan et al., 2005; Madras and Lapointe, 2008). Researchers have attempted to investigate the outcomes of these procedures systematically. However, integrating data across individual studies revealed too many inconsistencies for definitive conclusions to be drawn.
Ironically, since the tendency to treat OKCs more aggressively appeared to be reinforced by the recent WHO reclassification of OKC as a neoplasm, there have been increasing reports that OKCs can be treated for partial cure, or even complete cure, by simple marsupialization and decompression (Pogrel and Jordan, 2004; Maurette et al., 2006). The procedure involves creating a surgical window into the jaw, decompressing the intracystic high pressure, and leading to gradual reduction of the cyst size. Once again, these reports argue against the notion that OKC is a neoplasm, since typical neoplastic behavior is believed to be continued or persistent growth to occur after incomplete removal.
From a clinical point of view regarding the OKC, what really matters is not what it is called, but how it should be better managed. Recent studies related to the PTCH1 pathway have provided some insights into the development of molecular therapeutic approaches for Gorlin syndrome and its related sporadic tumors, including OKCs. The Hh pathway can be blocked at different levels, and Hh inhibitors could serve as attractive anti-tumor agents because of their specific effects on a small number of adult tissues (Pasca di Magliano and Hebrok, 2003). Several specific SMO inhibitors have been identified (Fig. 4C). Cyclopamine, a plant-based steroidal alkaloid, has been reported to inhibit cellular responses to Hh signal (Taipale et al., 2000). It specifically inhibits SMO activity by binding to its heptahelical bundle. Treatment of mice that carry Hh-dependent tumors with cyclopamine resulted in growth inhibition and regression of cancerous tissue, but did not affect the health of treated animals. Therefore, Hh inhibition causes few, if any, toxic effects on cells that do not depend on Hh signaling (Berman et al., 2002; Thayer et al., 2003). A novel small-molecule (Cur61414) inhibitor of the Hh pathway has been identified to block elevated Hh signaling activity resulting from oncogenic mutations in PTCH1. This small molecule can suppress proliferation and induce apoptosis of basaloid nests in the basal cell carcinoma model systems, but have no effect on normal skin cells (Williams et al., 2003). More recently, an orally active small molecule that targets the Hh pathway, GDC-0449, has also been reported to show anti-tumor activity in locally advanced or metastatic basal cell carcinoma in a phase 1 clinical trial (Von Hoff et al., 2009). In addition, several Hh-specific antagonists have been identified and tested. Inhibition of ligand activity has been reported with antibodies directed against SHH, which might be considered for treating tumors that are shown to require continuous ligand activity for survival (Watkins et al., 2003) (Fig. 4C). GLI can also be inhibited at the RNA level by targeting its transcripts with antisense oligonucleotides—an approach that has been used successfully in Xenopus (Pasca di Magliano and Hebrok, 2003). Zhang et al. (2006) postulated that antagonists of Hh signaling factors could effectively treat OKCs. Their suggested strategies include re-introduction of a wild-type form of PTCH1, inhibiting the SMO molecule by synthetic antagonists and suppressing the downstream transcription factors of the Hh pathway. They suggest that intracystic injection of a SMO protein-antagonist may be the most promising treatment option (Zhang et al., 2006).
Summary
The controversies over the nature of OKCs are in fact a reflection of our limited knowledge of the molecular basis of this fascinating entity. Analysis of the data accumulated so far appears to indicate that the p53 gene mutation is extremely rare in OKCs, although the level of p53 protein expression is heightened in its epithelium (Li et al., 1996; Gomes et al., 2009a). The association of OKCs with the inherited, autosomal-dominant, Gorlin syndrome appears to indicate pathogenic links or similarities in developmental pathways between isolated and syndrome-associated cases. Namely, PTCH1 mutations and misregulation of the Hh signaling pathway have a definite part to play in the development of OKCs, both in isolated cases and in syndrome patients (Li et al., 2008; Sun et al., 2008; Gomes et al., 2009a; Mendes et al., 2010; Pan et al., 2010). Despite all these recent advances, however, the evidence either supporting or refuting OKC as a cystic neoplasm is still insufficient for definitive conclusions to be drawn. The aggressive behavior and high recurrence rate of OKCs suggest a neoplastic potential and warrant special attention. Recent advances in genetic and molecular research have led to increased knowledge of OKC pathogenesis which hints at possible new treatment options. Since some investigators have advocated more conservative treatment for OKCs, namely marsupialization, future treatment strategy may even become molecular in nature and eventually reduce or eliminate the need for aggressive methods to manage the lesions. As a result of continuous efforts at understanding the molecular basis of this lesion, the various treatment protocols currently recommended will be better rationalized.
Footnotes
Acknowledgements
The author thanks Professor Tao Xu (Peking University School and Hospital of Stomatology, China) for his inspiring suggestions and recommendations. Studies performed in the author’s laboratory have been supported by research grants from the National Nature Science Foundation of China (30872900, 30625044, and 30572048). The author also thanks the following individuals for their contributions to the related research projects: Drs. Li-Sha Sun, Shuang Pan, Qing Dong, Xiao-Mei Gu, Jun-Wei Yuan, and Li-Li Xu.