
Abstract
Composite dental restorations represent a unique class of biomaterials with severe restrictions on biocompatibility, curing behavior, esthetics, and ultimate material properties. These materials are presently limited by shrinkage and polymerization-induced shrinkage stress, limited toughness, the presence of unreacted monomer that remains following the polymerization, and several other factors. Fortunately, these materials have been the focus of a great deal of research in recent years with the goal of improving restoration performance by changing the initiation system, monomers, and fillers and their coupling agents, and by developing novel polymerization strategies. Here, we review the general characteristics of the polymerization reaction and recent approaches that have been taken to improve composite restorative performance.
Introduction
Composite restorative materials represent one of the many successes of modern biomaterials research, since they replace biological tissue in both appearance and function. At least half of posterior direct restoration placements now rely on composite materials (Sadowsky, 2006). Unfortunately, demands on these restorations with regard to mechanical properties, placement, and need for in situ curing leave significant room for advancements, particularly with respect to their mechanical properties, polymerization shrinkage and polymerization-induced stress, thermal expansion mismatch, fracture, abrasion and wear resistance, marginal leakage, and toxicity (Anseth et al., 1995; Lovell et al., 2001a, b; Ferracane, 2005, 2008; Sadowsky, 2006). Ultimately, these shortcomings reduce a restoration’s lifetime and represent the driving force for improvement in dental composites. Clinical evaluations (Bernardo et al., 2007) and laboratory-based studies focused on composite durability (Drummond, 2008) also continue to highlight this need for new materials.
The development and implementation of composite dental restorative materials rely on a comprehensive understanding of each component of the composite and consideration of methods for changing each component. Here, we discuss basic components of composite restoratives and their role in the ultimate restoration. Composites are composed of three distinct phases, each with its own role in dictating material properties: the polymerizable resin, filler, and the filler-resin interface. The resin phase is composed of polymerizable monomers that convert from a liquid to a highly crosslinked polymer upon exposure to visible light, which catalyzes the formation of active centers, typically radicals, that induce polymerization. The filler has several roles, including enhancing modulus, radiopacity, altering thermal expansion behavior, and reducing polymerization shrinkage by reducing the resin fraction. Finally, the filler-resin interface serves as a bridge by coupling polymerizable moieties to the particle surface. Each component represents an opportunity for improvements in the overall composite and is the target of recent research reviewed here. Specifically, this article provides background for the general behavior observed in photopolymerization, including the origins of polymerization-induced shrinkage stress, photoinitiation systems used to improve the restoration curing behavior, recent research on these topics and novel monomers that have been explored, development of new photopolymerization mechanisms, and the filler and interface components of the formulation.
A 2001 review discussed development of polymeric composite restorative materials (Moszner and Salz, 2001). The article focused on methods for reducing polymerization shrinkage and achieving improvements in biocompatibility and wear resistance. Here, the focus is on providing general photopolymerization background and reviewing advances from the last five years. The vast research encompassed in this short time is a testament to the difficulty associated with successfully restoring tooth function and appearance and demonstrates continued room for advancement. We focus this review on recent research aimed at improving one or more attributes of dental restorative materials. Given the focus on recent research, only a very few of these developments have already been incorporated into clinical applications.
Photopolymerization of Multifunctional Monomers
Generally, the curing reaction in composite restorative materials involves visible-light-initiated photopolymerization of dimethacrylate monomers to form a highly crosslinked polymer. This photopolymerization reaction consists of three steps—initiation, propagation, and termination—and complexities arise in polymerization kinetics, network evolution, and the material property development (Kloosterboer, 1988; Anseth et al., 1995; Lovell et al., 2001a, b; Bowman and Kloxin, 2008).
With respect to the polymerization kinetics, both the propagation and termination reactions are diffusion-controlled. Even at low conversion (Anseth et al., 1994), the termination reaction, i.e., the coming together of two radicals that react to terminate each other, is diffusion-controlled and slowed by the network. Subsequently, the radical concentration increases and the observed polymerization rate also increases, a phenomenon referred to as ‘autoacceleration’. This process is important for dental composites, since it results in rapid curing on clinically acceptable time scales. In contrast to termination, the propagation reaction involves the reaction of a polymeric radical and a relatively mobile methacrylate moiety. This reaction’s nature is such that it does not become diffusion-controlled until significantly higher conversions, generally associated with the polymer becoming glassy, a process referred to as ‘vitrification’. As the polymer vitrifies, the propagation reaction slows and the polymerization ceases, i.e., autodeceleration occurs. This process is particularly important in dental composites, where autodeceleration results in residual, unreacted methacrylates that remain in the composite restoration.
To illustrate these points, we present the polymerization kinetic behavior of various methacrylate monomer formulations (Fig. 1a). Various dimethacrylate (bisphenol-A-dimethacrylate, BisGMA, and triethylene glycol dimethacrylate, TEGDMA) and monomethacrylate (isobornyl methacrylate, IBOMA, and tetrahydrofurfuryl methacrylate, THFMA) mixtures are polymerized, and the methacrylate conversion is plotted as a function of polymerization time. Mobility effects can be readily observed with increasing TEGDMA, where TEGDMA is a reactive diluent that reduces the viscosity, glass transition temperature, and network stiffness, and increases the maximum conversion that is achieved and the rate at which that conversion is achieved. The addition of IBOMA, a traditional monomethacrylate, slows the rate significantly, since its presence reduces the crosslinking density, which facilitates termination that lowers the polymerization rate. In contrast, the addition of THFMA, a novel monomethacrylate (see later section on Ultrarapid Monomethacrylates) that has been found to accelerate polymerization, accelerates the polymerization rate and increases the final conversion that is achieved. Though these systems were not polymerized at a clinically relevant irradiation intensity, the relative kinetics and conversions of the different systems can be more accurately measured and compared at this intensity.
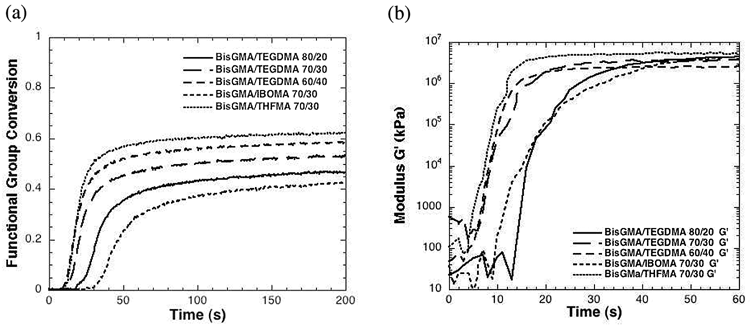
Polymerization kinetic behavior of various methacrylate monomer formulations.
In addition to complex polymerization kinetics, the polymer structure also evolves with numerous complexities. There are two critical, macroscopic demarcations that occur during polymerization. The first of these is the gel point conversion and represents the point at which an incipient gel is formed. In the chain-growth polymerization of dimethacrylates, this conversion is generally less than 5-10% and is critical for controlling the shrinkage stress (Kloosterboer, 1988). The second macroscale demarcation is the vitrification point, which represents the conversion at which the polymer becomes glassy, accompanied by a dramatic modulus increase. For the same formulations presented in Fig. 1a, Fig. 1b presents the storage modulus as a function of polymerization time. The initial modulus measurements exhibit significant error associated with the liquid nature of the resin, though once the system reaches and surpasses the gel point, the storage modulus increases by three to four orders of magnitude in a matter of only a few seconds.
Finally, in addition to the macroscopic observations of the network structure and material properties, it is important to understand that these polymer networks are extremely heterogeneous. This heterogeneity arises from two issues, the fact that microgels form near the sites of initiation, and the fact that pendant methacrylate groups in BisGMA/TEGDMA monomers and other analogous systems may be more reactive than their monomeric counterparts (Elliot et al., 2001). This heterogeneity has significant implications for composite restorative properties (Kannurpatti et al., 1998; Lovell et al., 1999a, b), contributing to the post-cure behavior noted for glassy methacrylate networks (Truffier-Boutry et al., 2006) and to the refractive index variation within the polymer matrix that enhances filler-induced translucency in composites (Howard et al., 2010).
Polymerization-induced Shrinkage Stress
Shrinkage stress is often considered the most significant problem with current restoratives and a primary contributor to premature failure in composite restorations, since it is capable of deforming tooth structures and causing microcracks and adhesive failure (Ferracane, 2005). The stress is dictated by a complex interplay among resin viscosity, volume shrinkage, polymerization rate, degree of conversion, modulus development, and network structural evolution, where each of these properties cannot be individually manipulated and studied without having a significant impact on other properties. A great deal of effort has been expended to develop novel monomers and fillers and distinct polymerization methods that reduce shrinkage stress while maintaining all other desirable material properties. Several generalized approaches have been attempted, focusing on (i) manipulating the curing protocol and timing to allow for relaxation and flow of the network prior to gelation, (ii) altering the amount of shrinkage that occurs through changes in the monomer or functional group density, and (iii) changing the polymerization mechanism from conventional radical chain-growth polymerization of methacrylates to alter the network structural evolution. These approaches are the overarching motivation for much of the research conducted in the dental restorative field.
Photoinitiation
Though non-photoinitiated polymerizations are still performed and studied (Achilias and Sideridou, 2004; Sideridou et al., 2008), photoinitiated polymerizations have enormous value in controlling the polymerization temporally, allowing adequate time for placement and manipulation of the restorative prior to curing. Clinically desirable visible light has reduced energy per photon relative to ultraviolet light, which limits the selection of suitable initiators and often necessitates multicomponent initiators. One such system, composed of camphorquinone and an amine, is well-known and is the most frequently utilized in dental materials. Continuing research focuses on understanding and optimization of initiator systems where the goals include enhanced initiation efficiency, improved coupling of the initiator absorption to the desired light source’s emission, reduction of colored by-products, and generation of new active center types, including cations for epoxy polymerization. Additionally, there remain concerns over the toxicity of the amine co-initiators that are used with camphorquinone. Ultimately, these needs drive the investigation of new photoinitiator systems.
Camphorquinone/Amine
The camphorquinone/amine initiation system continues to be the subject of research in efforts to determine optimum initiator and co-initiator concentrations for kinetics and polymer mechanical properties (Viljanen et al., 2005; Musanje et al., 2009). In this visible-light-activated initiator system, camphorquinone (CQ) absorbs a photon to generate a short-lived excited-state species that complexes with the tertiary amine to promote a sequential electron and proton transfer that creates the active α-aminoalkyl-initiating radical. Additional studies have evaluated alternative co-initiators to the commonly utilized ethyl-4-dimethylaminobenzoate (EDMAB), including N,N-dimethylaminobenzyl alcohol, 4-(N,N-dimethylamino)phenethyl alcohol (DMPOH), and N,N-3,5-tetramethylaniline (TMA) (Schroeder et al., 2007a, b, 2008; Schroeder and Vallo, 2007) and a variety of aliphatic and aromatic amines (Kim, 2005). With the goal of reducing or eliminating the amount of potentially toxic amine co-initiator, cyclic acetals and the naturally occurring 1,3-benzodioxole and its derivatives were evaluated as potential replacements for conventional amine co-initiators and were found to be effective co-initiators, resulting in kinetics and polymer properties similar to those of equivalent systems initiated by CQ/EDMAB (Liu et al., 2007; Shi and Nie, 2007; Shi et al., 2007). PPD (1-phenyl-1,2-propanedione) is a non-yellowing photosensitizer that has been proposed as an alternative to CQ (Ogunyinka et al., 2007; Schroeder et al., 2007a, b, 2008; Felipe et al., 2008; Schneider et al., 2009a,b), though it does not appear to be as efficient as CQ for visible light initiation.
One interesting approach for addressing amine co-initiator toxicity has been to develop polymerizable monomers that also function as co-initiators. Therein, the amine component is polymerized into the network via the methacrylate, which significantly limits the extractable amine. Several methacrylated amine co-initiators, such as ethylene glycol 3-diethylamino-propionate methacrylate, have been synthesized and demonstrated to exhibit polymerization kinetics and properties nearly equivalent to those of traditional BisGMA/TEGDMA systems with a CQ/EDMAB initiation system (Nie and Bowman, 2002; Wu et al., 2006; Wu and Nie, 2007).
Alternatives to Camphorquinone/Amine Systems
Phosphine oxide initiators absorb in the visible range and initiate via a cleavage mechanism that does not require a co-initiator. Phosphine oxide initiators exhibit minimal absorption beyond 420 nm and therefore are not ideal for use in dental material applications where the spectral output of LED curing lamps has been designed to match the absorption of CQ, which has an absorbance maximum at 468 nm. New initiation systems based on benzoylgermanium derivatives have been synthesized and demonstrated to be efficient visible light photoinitiators (Ganster et al., 2008a,b; Moszner et al., 2008b, 2009). Similar to phosphine oxides, the benzoylgermanium initiators undergo photodecomposition to form radicals without the need for a co-initiator. The benzoylgermanium initiators exhibit strong absorption up to 450 nm, which is advantageous for improved initiation efficiency in dental materials. The novel initiators were demonstrated to exhibit improved UV stability, comparable shelf stability, improved bleaching, and increased cure depths and polymerization rates relative to those of the CQ/amine systems.
Three-component photoinitiators composed of a photo-sensitizer dye, an electron donor photo-reductant, and an electron acceptor photo-oxidant offer enhanced efficiencies in the production of both free-radical and ionic species for photopolymerization. A variety of suitable dyes have been identified, which allow for variation in the active wavelengths that are considered, although dental materials applications continue to require the 400- to 500-nm region of the visible light spectrum. Amines are primarily used as electron donors, and diphenyl iodonium or triphenyl sulfonium salts are commonly used as electron acceptors, although several other examples of practical electron donors and acceptors are known. In (meth)acrylate-based radical photopolymerization, the addition of a diphenyl iodonium salt not only reduces the photon-wasting back electron transfer and recombination reactions, but also provides a means to recycle consumed photosensitizer, which effectively increases the photoinitiator concentration (Kim and Stansbury, 2009a,b). The introduction of a photo-oxidant component in a dental adhesive initiator system has been demonstrated to produce enhanced conversion in the presence of moisture (Guo et al., 2008), with analogous results shown in an unrelated system (Gomez et al., 2007). Similar three-component initiator systems are also used efficiently in cationically photocurable materials such as epoxides (Oxman et al., 2005; Crivello, 2009).
Soft-start Curing
One of the methods purported to reduce shrinkage stress without compromising other properties such as conversion or mechanical properties is to reduce the initiation rate with the use of lower irradiation intensities, i.e., soft-start curing. The soft-start curing method originated with work by Unterbrink and Muessner in 1994 (Unterbrink and Muessner, 1994, 1995) and continues to be the subject of significant study (Braga et al., 2005; Ferracane, 2005). Reduced irradiation intensity during the early stages of polymerization is hypothesized to allow stress relaxation to occur prior to vitrification. Soft-start curing has been adopted clinically and has undergone and continues to undergo significant research, due to the benefits of reducing shrinkage stress that might occur without compromising other properties (Braga and Ferracane, 2002; Lim et al., 2002; Witzel et al., 2005; Pfeifer et al., 2006; Cunha et al., 2007, 2008a,b). Though much of the work surrounding soft-start curing has taken conversion into consideration, a body of evidence supports the notion that reductions in shrinkage stress are often accompanied by subsequent reductions in conversion (Gonçalves et al., 2008; Pfeifer et al., 2008) and mechanical properties (Feng and Suh, 2006a,b). In addition to soft-start curing, Ferracane and co-workers (Musanje et al., 2009; Pfeifer et al., 2009b) have determined that changes to the photoinitiator composition and type also alter the polymerization kinetics, polymer structure, final conversion, and shrinkage stress, and that this factor must be considered in the design of an optimal photopolymerizable composite formulation. Frequently, shrinkage stress measurements are not coupled with in situ kinetic and functional group conversion results. It is inherently difficult to compare results across different specimens and experimental procedures when very small changes in conditions, shape, or conversion produce large changes in stress and material properties. Lovell et al. (2001b) demonstrated clearly both that small changes in conversion have a pronounced effect on properties, especially modulus, and that polymerization conditions had little effect on material properties other than through changes in the conversion.
Because of conversion’s importance in dictating material properties, the development of instrumentation that combines in situ conversion measurements by Fourier transform infrared (FTIR) spectroscopy with shrinkage or shrinkage stress measurements has enabled an effective means by which to study shrinkage and shrinkage stress (Lu et al., 2004a; Stansbury et al., 2005). These experiments demonstrated that the vast majority of stress development arises as the sample nears and ultimately undergoes vitrification. Therefore, stress relaxation can occur prior to vitrification; however, the magnitude of the stress reduction is minimal with respect to overall stress development (Lu et al., 2004a,b,c). These results also indicate that reduced stress that results from reduced irradiation intensity (soft-start/pulse-curing techniques) is often the result of changes in the conversion. In addition to the conversion, the sample geometry, initiation and polymerization rates, and curing protocols can also affect the stress that is achieved by altering the temperature profile that occurs as a result of the exothermic nature of the polymerization reaction. The BisGMA/TEGDMA (or similar) resin system has a limited ability to mitigate shrinkage stress without subsequent reductions in conversion and polymer properties. Extensive research has gone into and will continue to be devoted toward evaluating curing conditions and the subsequent effects on conversion, mechanical properties, and shrinkage stress.
Methacrylate Monomer Formulations
The resin phase of composite materials presently represents the area through which the greatest changes in composite practice may be achieved. That said, the requirements for the resin phase of the composite restorative are daunting: These materials must be stable on the shelf for years, then rapidly react to form a highly crosslinked polymer with high modulus, high hardness, and high glass transition temperature while matching the thermal expansion of the tooth, limiting extractables, minimizing moisture uptake, being chemically inert, and having minimal shrinkage and shrinkage stress. Clearly, while the potential improvements in this area are dramatic and the array of possibilities for replacement is also vast, only through careful and insightful manipulation of the current resin phase will significant results be realized.
Currently, methacrylate resin formulations dominate both the commercial market and research evaluation. The resin phase is composed primarily of dimethacrylate monomers typically selected from BisGMA, BisEMA, and/or UDMA. These base monomers result in restorative materials with excellent mechanical properties, rapid polymerization, and low shrinkage. However, resins composed of monomers such as these generally result in low methacrylate conversion, which leads to significant amounts of unreacted monomer that may be leached from the restoration over time, resulting in concerns regarding long-term biocompatibility. High resin viscosity also limits the ability to incorporate high filler volumes into the resin. To achieve low volume shrinkage and high mechanical properties such as modulus and wear resistance, filler contents of 60 to 87 wt% (Lohbauer et al., 2006) are necessary. To reduce viscosity, enabling high filler content to be incorporated, low-viscosity reactive diluents, most commonly TEGDMA, are used. The inclusion of a reactive diluent reduces viscosity and increases conversion, but also generally reduces the modulus and results in higher volume shrinkage and shrinkage stress. Both the resin formulation and filler type and content affect the final composite curing and mechanical properties. Dental restorative material compositions of dimethacrylates are a balance between the relative amounts of base materials and reactive diluents, which results in trade-offs among resin viscosity, polymer properties, and monomer conversion. It is generally not practical to study simultaneously the effects of alterations to the resin formulation and the effects of fillers on new resin compositions. Therefore, new resin formulations are most commonly studied without the addition of fillers, and subsequent studies on optimal resin candidates evaluate the synergistic effects of changes to both components.
Formulations containing one or more of the base monomers, BisGMA, EBPADMA, UDMA, and TEGDMA, have been utilized commercially for decades. The monomer interactions, polymerization kinetics, and polymer properties resulting from these materials are complex. Numerous investigations are being and have been conducted to evaluate various combinations of these dimethacrylate materials, in efforts to understand the interrelationships among composition, resin viscosity, conversion, shrinkage, flexural strength, fracture toughness, water sorption and solubility, and rheology (Sideridou et al., 2003; Marcinkowska and Andrzejewska, 2006; Charton et al., 2007; Ellakwa et al., 2007; Gonçalves et al., 2008, 2009; Pfeifer et al., 2009a). In addition to formulations containing only the traditional base monomers, a vast amount of research has focused on developing systems with alterations and improvements to these formulations, including the incorporation of monomethacrylate diluents, dimethacrylates, and multimethacrylates. Changes to the polymerization mechanism, which also involves changes to the monomers, are reviewed in a later section. An array of monomer structures for the base dimethacrylate materials as well as new monomers are given in Table 1.
Several representative methacrylate monomer structures utilized in resin based composite restorative materials.
Multimethacrylates
Because the conventional dimethacrylate monomers have worked well in many regards, one target of current research is to select, synthesize, and evaluate dimethacrylates that preserve the desirable attributes of the conventional restoratives while simultaneously addressing their shortcomings. As such, a wide range of dimethacrylate monomers has been synthesized and evaluated as potential restorative materials. These new monomer systems show promise for maintaining or improving properties such as conversion, water sorption, volume shrinkage, and shrinkage stress, with the overarching goal of creating a restorative material with improved performance and service lifetime.
The use of a bisphenol-A core, as is contained in BisGMA- and EBPADMA-based materials, provides both high strength and toughness to the resulting polymers. Hence, the development of dimethacrylate derivatives of bisphenol-A has been an active research area, where modifications of bisphenol-A-based dimethacrylate systems have included the use of pendant bulky (aromatic) constituents (Ge et al., 2005) as well as pendant alkyl urethanes (Khatri et al., 2003) to increase molecular weight and thereby decrease volume shrinkage. Oligomeric bisphenol-A monomers have also been modified with pendant urethane acrylates and exhibited reduced volume shrinkage and improved mechanical properties (Chen et al., 2008). Increasing the ethylene oxide chain length between the bisphenol-A core and the methacrylate functional group reduces viscosity and increases conversion, but also increases water sorption and decreases flexural strength (Ogliari et al., 2008). Incorporating methylated and fluorinated derivatives of BisGMA into BisGMA/TEGDMA resins has resulted in greater hydrophobicity and reduced water sorption (Pereira et al., 2007), but no significant improvements in mechanical properties (Pereira et al., 2005, 2007).
Recent developments regarding public perceptions of bisphenol-A toxicity may have a strong influence on steering future monomer development efforts toward bisphenol-A alternatives. Numerous other methacrylate monomer types are already being developed and evaluated that would achieve this end. Derivatives of urethane dimethacrylate have been synthesized to increase molecular weight, reduce water sorption, and/or increase mechanical properties by incorporating aromatic or aliphatic groups (Atai et al., 2007; Moszner et al., 2008a; Kerby et al., 2009). Bile acids were utilized as starting materials to form multimethacrylate monomers (Gauthier et al., 2009). These materials showed reduced volume shrinkage and promising mechanical properties; however, they exhibited extremely high viscosities (higher than BisGMA). Polyhedral oligomeric silsesquioxane methacrylates (POSS-MA) were evaluated as alternatives to BisGMA (Fong et al., 2005), and it was found that small amounts of POSS-MA (2-10 wt%) did indeed improve the mechanical properties of these resins, while dimethacrylates based on cycloaliphatic epoxides showed kinetics and mechanical properties comparable with those of BisGMA (Shi and Nie, 2008). Methacrylated beta-cyclodextrin derivatives have also been evaluated as alternatives to BisGMA and were found to exhibit flexural strength and volume shrinkage comparable with those of BisGMA/TEGDMA (Hussain et al., 2005).
Many previously developed dimethacrylate materials exhibit excellent properties with regard to modulus, water sorption, conversion, and so forth. When one considers that a dental restorative material must balance numerous properties, the dimethacrylate materials to date generally result in trade-offs among resin viscosity, polymer properties, and monomer conversion. For example, monomers with higher molecular weights often result in low shrinkage and excellent material properties, but also concomitant increases in resin viscosity (limiting the incorporation of filler) and reduced ultimate conversion. Hence, while numerous dimethacrylate derivatives have shown great promise as dental restorative materials, improvements in overall properties relative to BisGMA/TEGDMA are generally modest, hence the limited commercial incorporation of these monomers.
Ultrarapid Monomethacrylates
For many years, the inclusion of monomethacrylate monomers in dental resins was rightly seen as problematic, since inclusion of traditional monomers of this type generally slows the polymerization, reduces the modulus and crosslinking density, increases the shrinkage, and leads to an increased amount of extractable materials. The paradigm for inclusion of monovinyl monomers into dental resins was changed with the development by Decker of a novel class of monovinyl (meth)acrylate monomers that exhibited greatly enhanced polymerization kinetics and significantly improved mechanical properties. These monomers contain secondary and tertiary functionalities such as urethanes, carbonates, cyclic acetal, morpholine, cyclic carbonates, hydroxy/carboxy, oxazolidones, and aromatic rings (Decker and Moussa, 1990, 1991a,b; Jansen et al., 2003; Berchtold et al., 2004, 2008, 2009; Lu et al., 2005; Kilambi et al., 2007a,b, 2009) that lead to their unique polymerization and polymer property behavior. These monomers exhibit rapid polymerizations that rival and often exceed those of equivalent di(meth)acrylates, and the polymers exhibit a high glass transition temperature. Additionally, the mono(meth)acrylates exhibit high conversion, limiting the potential for leachable monomer. These materials showed great promise when utilized as diluents, and several monomethacrylates were evaluated as alternatives to TEGDMA (Lu et al., 2005; Kilambi et al., 2009). For example, morpholine carbonyl methacrylate with BisGMA exhibited 21% increased conversion, 3.5 times faster polymerization rate, and 30% reduced volume shrinkage as a resin system (Lu et al., 2005) and 13% increased conversion, 3 times faster polymerization rate, and equivalent or improved mechanical properties as a composite system relative to a control BisGMA/TEGDMA system (Kilambi et al., 2009).
Acidic Monomers
Recent developments in methacrylate resins have investigated the incorporation of acidic functional groups into the monomer structure. Incorporating acidic monomers in relatively small mole fractions into methacrylate resins may enable a separate adhesive layer to be eliminated and result in improved overall performance. Current acidic resins do not exhibit the necessary mechanical properties to function as resin-based composites, and hence research has focused on developing acidic monomers with improved mechanical properties. Acidic monomers have been synthesized from o-hydroxyaryl phosphonates that exhibited rapid polymerization kinetics (Sahin et al., 2006). Bisphenol-A derivatives have been produced both with carboxylic acid and phosphonic acid functional groups without degradable esters (Sahin et al., 2009) and with carboxylic, amide, and hydroxy functional groups to improve adhesion (Yagci et al., 2006). Other derivatives of BisGMA, including carboxylic acid functionalized monomers, exhibit comparable mechanical properties but increased water sorption (Atai et al., 2004). The increased moisture absorption associated with the acid is a common problem, since the presence of the acidic group increases the hydrophilicity of the material through increased polarity and charge density. Acidic monomer-containing composites with various aromatic core structures were evaluated for mechanical properties and found to exhibit properties similar to those of BisGMA/TEGDMA (López-Suevos and Dickens, 2008). The use of acidic monomers necessitates consideration of hydrolytic stability. Acrylamides exhibit increased hydrolytic stability compared with esters, and especially when utilized under acidic conditions, acrylamides present a promising alternative to methacrylates. Bis-acrylamides showed similar reactivity, flexural strength, and flexural modulus when compared with similar dimethacrylate resins (Moszner et al., 2006b). The use of internal amide functional groups has also been considered. These monomers are derivatives of bisphenol-A and exhibited significant reductions in volume shrinkage, but also resulted in increased viscosity and, due to low solubility, are able to be incorporated only at levels up to 5 mol% in BisGMA/TEGDMA resins (Yagci et al., 2006).
Novel Polymerization Mechanisms
Conventional radical-mediated chain-growth polymerization of dimethacrylates has found incredible utility in composite restoratives; however, it is fundamentally limited in several aspects. The chain-growth polymerization mechanism leads to early gelation (Kloosterboer, 1988), while the methacrylate consumption is linked to a defined volume reduction associated with the consumption of each methacrylate (Patel et al., 1987). Improvements in the methacrylate monomer structure, as noted previously, have the potential for addressing many of the shortcomings of current composites; however, an even greater potential lies in completely changing the reaction mechanism, either by changing the active center (from radical to cationic), by changing the nature of the network/molecular-weight evolution (by changing to a step growth reaction or by changing to a covalent adaptable network), by changing the nature of the reactive chemistry (by going to ring-opening species), or by changing the physical behavior that arises during polymerization (by inducing phase separation). Exciting research has focused on bringing each of these developments to dental restorative materials, and their efforts are summarized here.
Polymerization-induced Phase Separation
Incomplete polymerization, volumetric shrinkage, and stress are among the primary disadvantages of current resin-based dental composites. Generally, attempts to increase double-bond conversion exacerbate polymerization shrinkage and stress. In one creative approach, specific methacrylate monomers, chosen to be miscible as liquids but phase-separated at higher conversions, were incorporated into conventional methacrylate resins and composites. When phase separation occurs, the volume expands, eliminating a portion of the volume shrinkage that arises from the methacrylate polymerization.
In particular, the use of dimer-acid-derived dimethacrylate (DADMA) monomers in novel dental resin formulations is examined as a potential means to address the combined aims of high conversion and low shrinkage and shrinkage stress. A series of DADMA monomers, with various connecting groups used to attach the C36 dimer acid core to the methacrylates, were formulated as reactive diluents with BisGMA, UDMA, and/or BisEMA to manipulate comonomer compatibility and polymeric properties. The dimer-acid-derived monomers, DADMA I, II, and III (Table 2), were prepared as previously described (Trujillo-Lemon et al., 2006). The DADMA monomers have molecular weights of 673-849 g/mol, with initial methacrylate group concentrations of 2.4-2.7 mol/L as compared with the values for TEGDMA of 286 g/mol and 7.5 mol/L, respectively. Monomer I has no hydrogen bond donor functionality and presents the lowest viscosity of the DADMA series. It has limited miscibility with BisGMA or UDMA, but is compatible with BisEMA. Conversely, DADMA monomers II and III, which do form hydrogen bonds through the hydroxyl or urethane functionality, respectively, are compatible with BisGMA or UDMA, but are only partially miscible with BisEMA. Therefore, ternary compositions that combined one DADMA monomer with either BisGMA or UDMA and BisEMA allowed formulations to be prepared in which the degree of thermodynamic compatibility could be precisely tuned. Formulations near the thermodynamic stability boundary, as monomers, underwent polymerization-induced phase separation (PIPS) to generate heterogeneous polymers. The extent of phase separation depends on the resin composition and reaction conditions, with greater sensitivity apparently related to reaction temperature as opposed to reaction kinetics.
Polymer Properties of Ternary Dimer Acid Dimethacrylate-based Resins and Monomer Structures of Various Dimer Acid Dimethacrylate Dimethacrylates.
Uniquely, methacrylate conversion values of all the experimental resins were significantly greater than those of the control (Table 2), while in appropriate formulations, the flexural strength of the experimental resins was equivalent to that of the BisGMA/TEGDMA control. Despite their higher conversion values, the polymerization shrinkage results for all the experimental formulations were well below those of the control, with several experimental materials demonstrating a modest volume recovery during the post-cure observation interval. This type of novel behavior has been reported for other systems involving PIPS associated with pre-polymer additives (Velazquez et al., 2004; Schroeder et al., 2007a). In an analogous fashion, the experimental resins display lower polymerization shrinkage stress compared with the control (unpublished observations).
Thiol-Ene Photopolymerization
Work has also focused on utilizing the thiol-ene photopolymerization mechanism as a means for circumventing the problems with conventional methacrylate polymerization. The thiol-ene polymerization reaction is ideally suited for dental restorative materials, since these reactions are rapid photopolymerizations that achieve high functional group conversion, are not inhibited by oxygen, and proceed via a step-growth polymerization mechanism in which propagation and chain transfer alternate (Cramer and Bowman, 2001; Hoyle et al., 2004, 2010; Lu et al., 2005; Hoyle and Bowman, 2010). The step-growth nature of the polymerization results in uniform polymer networks with narrow glass transition regions and reduced brittleness. Also, the gel-point conversion is significantly higher in thiol-ene networks as compared with methacrylate networks, because of the step-growth polymerization. Thus, shrinkage that occurs before gelation, which now represents a large fraction of the total shrinkage, can be accommodated by flow rather than stress evolution, and hence thiol-ene systems exhibit significant reductions in polymerization shrinkage stress (Carioscia et al., 2005; Lu et al., 2005; Cramer et al., 2010). Though thiol-ene systems exhibit a number of very attractive properties, including high glass transition temperature, results to date have demonstrated that they also exhibit reduced flexural modulus and strength relative to BisGMA/TEGDMA controls (Carioscia et al., 2005, 2007; Lu et al., 2005; Fairbanks et al., 2009; Cramer et al., 2010).
Utilizing thiol-ene systems in combination with methacrylate systems in methacrylate-thiol-ene systems is one method to combine the advantages of both the thiol-ene and methacrylate systems. Methacrylate-thiol-ene systems were demonstrated to exhibit cure time and flexural modulus and strength equivalent to those of BisGMA/TEGDMA, while achieving increased levels of conversion and exhibiting dramatic reductions in shrinkage stress (Cramer et al., 2010). In fact, reductions in shrinkage stress in methacrylate-thiol-ene formulations are greater than for bulk thiol-ene systems. The increased reduction in shrinkage stress is due to the hybrid nature of the polymerization. The reaction often proceeds in two relatively distinct stages. The first stage is dominated by methacrylate homopolymerization with chain transfer to thiol, while the second stage is dominated by thiol-ene polymerization (Lee et al., 2007a,b).
One route to alleviate stress in polymer networks, already demonstrated for thiol-ene systems but also applicable to methacrylates, is the creation of a covalent adaptable network in which the bond structure of the network remains covalent, yet each individual bond can be broken and re-formed (Scott et al., 2005; Kloxin et al., 2010). In dental materials, this outcome is achieved by the incorporation of allyl sulfide moieties into multifunctional monomers that underwent a thiol-ene photopolymerization. While the polymerization proceeds, the allyl sulfide participates in an addition-fragmentation process that allows the forming polymer network to relax throughout the polymerization rather than simply prior to gelation. This network adaptation process has been shown to enable polymerization shrinkage stress reductions of up to 75% when compared with otherwise identical monomers in which the allyl sulfide is replaced with a propyl sulfide (Kloxin et al., 2009).
Hybrid Polymerization Reactions
Hybrid polymers are formed from comonomers with different reactive groups that polymerize via different curing mechanisms and are often utilized to synergistically achieve desired properties. Typically, polymerizations occur in parallel to form an interpenetrating polymer network (IPN) or, more generally, a material that is formed from two distinct polymerizations that generally have significant bonds between the two materials. For example, the use of methacrylate/vinyl ether systems facilitates sequential one-step hybrid polymerizations (Lin and Stansbury, 2003, 2005) where the methacrylate polymerizes via a radical reaction and the vinyl ether polymerizes via a cationic mechanism. The order of the reactions can be controlled by the selective addition of inhibitors of each polymerization type or through manipulation of the initiating wavelength-initiator combination. Here, the incorporation of vinyl ether monomers is desirable, since they exhibit high reactivity, no oxygen inhibition, and low toxicity and irritation properties. An additional benefit of hybrid polymerization is that reduced shrinkage stress can often be demonstrated, particularly when one polymerization type largely precedes the second polymerization (Carioscia et al., 2007; Lee et al., 2007b). In this manner, the gel-point of the entire system is delayed, allowing flow and relaxation to occur and accommodating any shrinkage without stress generation during the intervening period.
Ring-opening Polymerization
The implementation of ring-opening polymerization in dental restoratives has long been sought, even going back to Bowen’s original work (Bowen, 1956), for many reasons. The primary reason that ring-opening polymerization has received attention is underpinned by the unique shrinkage behavior observed in these polymerizations. Whereas methacrylate (and thiol-ene) photopolymerizations involve the conversion of a carbon-carbon double bond into single bonds, the ring-opening reaction relies on the opening of a cyclic structure to facilitate intermonomer bonding and crosslinking. Inherent to the cyclic structures is that significantly less volume shrinkage occurs when rings are opened.
A recent exciting development in ring-opening polymerization is the commercial release of the cationically photopolymerizable silorane material (Filtek LS) by 3M/ESPE (Weinmann et al., 2005). The resin chemistry relies on the ring-opening polymerization of a combination of proprietary and readily available cycloaliphatic monomers. The silorane terminology derives from the novel monomer composed of a cyclic siloxane core appended with 4 oxirane reactive groups. The cyclohexene oxide-type oxirane rings are significantly more reactive compared with a simple epoxy analog, and the very hydrophobic siloxane structure effectively balances the potential hydrophilicity of the polyether backbone generated by the ring-opening polymerization. The cationic reaction is activated by a visible-light photoinitiator system with camphorquinone as photosensitizer, a tertiary aromatic amine as a photoreductant, and an iodonium salt as an electron donor that creates the active cationic species. The filler selection, the filler surface treatment, and the adhesive required for bonding the composite to dentin and enamel are also designed to accommodate the cationic curing process. A successful, cationically initiated dental restorative material certainly represents a significant challenge, as implied by the fact that Bowen initially tried and abandoned cationic epoxide materials before developing BisGMA in his seminal work on dental restorative materials (Bowen, 1956). The available literature on silorane physical and mechanical properties has recently been reviewed (Duarte et al., 2009). A mechanical property characterization targeting several different length scales has been conducted with the silorane composite material along with several commercial composites based on conventional dimethacrylate resin chemistry (Ilie et al., 2009). The cationic silorane and free-radical methacrylate materials provided comparable properties initially and after water storage, with the silorane demonstrating a better retention of initial mechanical properties during extended storage in alcohol. The oxirane ring-opening polymerization process is quite exothermic, since it relies on the relief of the substantial ring strain to provide the driving force for the polymerization process. The advantage of low polymerization shrinkage associated with ring-opening polymerization arises due to an inherently lower molar shrinkage coefficient, as well as greater concentrations of chain ends in comparison with methacrylate polymerizations.
Monomers containing suitably placed cyclopropyl groups capable of undergoing ring opening based on free-radical re-arrangement mechanisms have been developed for dental applications (Moszner et al., 2006a; Moszner and Salz, 2007). Here, a vinyl group next to the highly strained bicyclic structure facilitates the re-arrangement that generates a single lower-energy ring. Based on this approach, experimental dental composites that involved free-radical copolymerization of the ring-opening monomers with conventional dimethacrylates were evaluated to demonstrate that composites with good mechanical strength and much-reduced polymerization shrinkage compared with control materials were available. Whereas the polymerization of monomers with strained small ring structures is driven primarily by enthalpy change, the ring-opening polymerization of monomers with larger ring structures is also possible based on entropy effects. Several bicyclic monomers that engage in either radical or cationic double-ring-opening polymerization have been developed and examined for their potential to reduce polymerization shrinkage in dental materials (Miller et al., 2005; Moon et al., 2005; Chappelow et al., 2008). A detailed mechanistic study of 2-methylene-7-phenyl-1,4,6,9-tetraoxaspiro[4.4]nonane, a bicyclic monomer known to provide ring-opened polymer, indicated significant issues, including susceptibility to moisture-induced side-reactions in cationic and radical-assisted cationic polymerizations (Ge et al., 2006). It is worth noting that new reaction mechanisms, other than free-radical methacrylate, must also consider stable adhesion between the restoration and the tooth.
Fillers and Filler Modifications
In addition to research on the photoinitiation process and the monomers used, research on fillers constitutes a large potential source of improvement in composite-based dental restoratives. In fact, a significant fraction of the practically implemented improvements in composites in recent decades has occurred in the nature, type, size distribution, and surface modification of the filler. An excellent review (Klapdohr and Moszner, 2005) focused on the inorganic filler component of dental composites and related filler composition, morphology, and loading content with properties conveyed to composites. This review also examined a variety of silane surface modifications and sol-gel-based hybrid inorganic/organic materials. Here, we survey continued developments related to filled dental polymers, including several recent approaches that involve the analytical characterization of composite materials as well as the implementation of advances in filler technology that result in improved composite restoratives.
Nanofillers in Dental Composites
Significant attention has been devoted to nanofilled materials, including improvements realized by the incorporation of nanofillers into commercial composite materials and research aimed at the development of new nanofillers. A recent review focused on nanofilled dental composite materials (Soh et al., 2006), and a separate report centered on how nanofillers affect composite mechanical properties and behave distinctly differently compared with micro- or macro-scale fillers (Crosby and Lee, 2007). Nano-sized fillers can be categorized as either isolated discrete particles, with dimensions of approximately 5 to 100 nm, or fused aggregates of primary nanoparticles, where the cluster size may significantly exceed 100 nm. The enormous rise in filler surface area and the corresponding thickening effect on composite paste consistency associated with decreasing filler size limit the content of discrete nanoparticles to relatively low loading levels, whereas high contents of nanoparticle clusters are manageable with appropriate surface treatment. A spatially resolved nanoindentation study examined Filtek Supreme XT (A3 Dentin) as a nanofilled composite and demonstrated significant differences in the dynamic complex modulus as a function of positioning within the matrix, within a filler cluster, or at the matrix-filler interface (Ilie et al., 2009). A study on the influence of mono-, bi-, and tri-modal distributions of fillers on the wear properties of composites showed that filler size and shape significantly influence wear resistance, with the inclusion of nano-sized filler a critical feature, often leading to enhanced properties (Turssi et al., 2005). A similar dependence of toothbrush abrasion resistance on the presence of nanoparticles in commercial dental composites has been shown (Cavalcante et al., 2009).
In terms of novel nanofillers, a variety of sol-gel-derived hybrid organic/inorganic monomers (ormocers) as well as functional silanes were described in a review of their design for dental composite applications (Klapdohr and Moszner, 2005). A systematic study of the sol-gel synthetic approach was used to produce nearly monodisperse silica particles of adjustable size from 5 to 450 nm. Silanization conditions were identified that yield uniform surface coverage regardless of particle size, and dental composites were formulated by incorporation of the nanoparticles alone or in combination with a barium glass filler so that particle dispersion and resin/filler adhesion potential could be examined (Kim et al., 2007). The introduction of bonded or non-bonded nanofiller in a hybrid composite was evaluated in terms of its effect on abrasion and attrition wear of the composite. While the use of non-bonded nanofiller does provide a means to reduce polymerization shrinkage stress in dental composites, it may also reduce wear resistance. Resin viscosity was a cofactor with composites based on the lower-viscosity resins, which achieve higher degrees of conversion, performing better in wear studies than higher-viscosity resins with the same filler (Musanje and Darvell, 2006). The presence of low to moderate amounts of Montmorillonite clays as a nano-scale layered silicate filler in a model dental resin was examined, with attention given to dispersion and exfoliation potentials dependent on loading level and polarity (Discacciati and Orefice, 2007). A method was developed to prepare single-walled carbon nanotubes (SWCNT) with suitable compatibility and polymerizability such that they could be introduced into dental composites as a secondary filler. The SWCNT were oxidized and then silanated in two steps to attach polymerizable surface groups. Addition of small amounts of the carbon nanotube filler to a commercial composite (Durafill) yielded a material with compromised esthetics, but one that was still photocurable. A good dispersion of the SWCNT in the composite was demonstrated, along with a significant increase in flexural strength compared with that of the unaltered composite material (Zhang et al., 2008).
Additional Dental Fillers and Composite Performance
The use of mesoporous silica fillers has been suggested as a means to eliminate the silane-mediated interface between filler and matrix, while providing a potentially more stable direct mechanical interlocking. A study evaluating the use of silica particles with interconnected pore structures as well as a non-porous silanized silica filler showed that optimized filler loading and mechanical reinforcement were achieved with a mixture of the two fillers (Samuel et al., 2009). The potential anisotropic effects of fiber-based fillers on polymerization shrinkage have also been demonstrated (Tezvergil et al., 2006), as well as the interactions between fibrous and particulate fillers in complex composite materials (Garoushi et al., 2008). Electrospun continuous nano-fiber-reinforced dental polymers have also been evaluated, with a focus on the fiber-matrix interface being a critical feature (Gao et al., 2008; Lin et al., 2008). A composite wear study with bioactive glass-ceramic fillers was conducted, and it demonstrated improved performance at low levels of surface porosity, but reduced wear resistance in the case of highly porous filler surfaces (Tan et al., 2010). An examination of nano-fibrillar silicate crystals showed the potential for improved mechanical properties, but also highlighted the difficulties of obtaining uniform dispersion of the nano-structures in the matrix (Tian et al., 2008). The combination of calcium phosphate nanoparticles with silicon nitride whiskers produced a composite material with caries inhibition potential and good mechanical properties (Xu et al., 2009). Other composites designed to promote remineralization were also examined (Mehdawi et al., 2009).
The influence of filler particles on the rheology and handling properties of dental composites has been extensively evaluated. Filler incorporation converts the Newtonian behavior of unfilled resins to composites that exhibit shear-thinning and thixotropic behavior, with micro-sized fillers inducing relatively subtle effects compared with the dramatic changes associated with nanofillers (Lee and Bowman, 2006; Beun et al., 2009). One investigation of commercial composite materials used an oscillatory compressive rheometer to highlight the substantial differences in viscoelastic behavior of these uncured composite pastes (IB Lee et al., 2007). Additional studies correlating the rheological properties of commercial composites with their time-dependent slumping resistance have been conducted (Lee et al., 2008). Related to this, a method to quantify the effects of particle size and morphology on handling properties of experimental composites was developed. The maximum force and work involved with the separation of a probe from uncured paste are related to the ‘stickiness’ of the composite, with differences noted between spherical and irregular particulate fillers (Kaleem et al., 2009).
Silane Treatment of Inorganic Fillers
Surface modification of most fillers used in dental composites is necessary: (a) to reduce the filler surface energy such that composite paste consistency and hydrophilicity are reduced while filler dispersion within the resin is enhanced; and (b) to provide a functional interface that permits covalent attachment between the polymer matrix and the reinforcing higher-modulus filler. Hydrolyzed trialkyloxysilane groups of the coupling agent react and interact with surface silanols (or other groups, depending on the filler composition) as well as with themselves to form an imperfect array of covalent and hydrogen-bonded attachment sites that generally yield a dense, multi-layered interface. While methacryloxypropyltrimethoxysilane (MPS) remains the most widely used surface treatment for the inorganic fillers used in dental composites, several new studies related to either MPS or alternative silane surface modifiers have also been reported. A solid-state NMR-based analysis of MPS-treated fillers used in model composite materials suggests that the primary silane attachment mainly involves –CH2SiO2(OH) (T2) structures and also used FTIR spectroscopy to verify the conversion restrictions imposed by fillers in composite materials (Nunes et al., 2008). A study of silane-mediated bonding between a resin composite cement and silica-coated titanium evaluated the efficacy of a variety of functional silanes, including several lacking (meth)acrylate-reactive sites. This result verified the advantages of MPS and its acrylate analog, but also pointed out the potential of a mercapto-functionalized silane to serve as a durable coupling agent (Matinlinna et al., 2007). In contrast, the addition of mercapto-functionalized silica nanoparticles to a thiol-ene-based resin was examined for its effects on photopolymerization reaction kinetics. The presence of a stoichiometric thiol to ene imbalance in the vicinity of the nanoparticle interface resulted in a suppression of the overall thiol-ene reaction rate at low filler loading, but at high nanoparticle content, viscosity effects associated with the particles produced an enhanced photopolymerization rate (Lee and Bowman, 2006).
A neutron scattering analysis of silanized nanoparticles over a range of loading levels in a model dental resin showed that dispersion was enhanced, although still not fully achieved, with an MPS surface treatment rather than n-octyltrimethoxysilane at the interface (Wilson et al., 2007). The effect of the silane structure applied to a filler surface on the handling properties of uncured composite paste as well as the photopolymerization process and the final composite properties were investigated (Wilson and Antonucci, 2006). The potential benefits of dual silane treatments were considered where the combination of a styryl functionalized silane with MPS demonstrated a higher modulus in the photocured composite compared with an MPS-treated control composite, while the combination of MPS with the non-functional n-octyltrimethoxysilane improved the handling properties of the composite paste without reducing the mechanical properties of the cured material. In another study, allyltriethoxysilane was used to surface-treat titanium dioxide nanoparticles (< 20 nm). The modified TiO2 particles were dispersed in a commercial composite (Z100) at 1 wt%, with a significant increase in hardness and flexural strength observed compared with the unaltered control (Xia et al., 2008). The degree of spatial ordering of a 500-nm spherical silica filler in triethylene glycol dimethacrylate could be enhanced with MPS-treated filler or disrupted by the introduction of dimethylaminoethyl methacrylate with untreated filler. Compressive testing demonstrated an improved mechanical strength in the ordered materials (Wan et al., 2008). The wear resistance of experimental hybrid composites prepared with filler pre-treated with either MPS, MPS combined with a non-functional fluorinated silane, or a novel aromatic methacrylate-functionalized silane was studied. The results showed that the new functional aromatic silane has excellent potential as a hydrophobic, matrix-resin-compatible coupling agent in terms of wear, in comparison with MPS surface-treated filler (Nihei et al., 2008). The structures of various silane modifiers for fillers are summarized in Fig. 2. Surface treatment of particulate and short-fiber fillers based on ultra-high-molecular-weight polyethylene (3-6 × 106 g/mol) demonstrated improved composite toughness, due to the formation of a ductile interface; however, composite strength and modulus were compromised (Ranade et al., 2006). Ultimately, filler shape and surface modification relative to the resin dictate the effects of the filler on the composite properties.
Various silane modifiers used to induce enhanced dispersion, coupling, and copolymerization in filled composite restorative materials.
These effects of the filler on composite properties are prominent in several investigators’ work, where it has been demonstrated that the methacrylate conversion in composite materials is inversely related to the filler loading level (Halvorson et al., 2003; Tanimoto et al., 2005; Garoushi et al., 2008; Nunes et al., 2008). One study focused on the effect of varied MPS coverage (from 0 to 20 wt%), with the finding that increased silane surface treatment on 0.6 µm zirconia glass filler resulted in lower overall conversion, based on relatively poor reactivity of methacrylate groups within the mobility-restricted interfacial silane layer (Halvorson et al., 2003). Contrary to this, a separate study indicated no significant difference in conversion of composites prepared with OX50 silica nanoparticles treated with MPS (from 1 to 10 wt%), although spectroscopic evidence of variations in silane orientation dependent on concentration was provided (Sideridou and Karabela, 2009). Related to the observations of reduced conversion in filled resins, several studies concerned with the light attenuation based on absorbance and scattering effects in composites have been presented (Emami et al., 2005; Musanje and Darvell, 2006; Arikawa et al., 2007; Shortall et al., 2008; Howard et al., 2010). Clearly, a combination of light attenuation associated with increased scattering as well as decreased mobility in the interfacial layer may be responsible for the observed relationship between filler loading and methacrylate conversion, depending on the physical and chemical attributes of the sample.
New Analytical Approaches Applied to Filled Materials
One significant need in the composite restorative field relates to the development of new techniques for determining both the properties of filled systems and the filler’s impact on material properties. In this area, several analytical techniques have recently been developed or newly applied to help characterize dental composite materials. In one exciting approach with the potential for rapid evaluation of a wide spectrum of materials, a combinatorial two-dimensional array was applied to composite specimens with discrete variations in filler composition as well as continuous gradient variations in the methacrylate conversion (Lin-Gibson et al., 2009). Different proportions of a 0.7-µm silanized filler were evaluated with or without the presence of a small amount of nanofiller. A rastering nanoindentation technique was used to obtain the localized hardness and elastic modulus and to assess the viscoelastic response based on a progressive load scratch test. The filler loading and the presence of nanofiller were found to alter the viscoelastic behavior of the composite materials as well as macrophage cell adhesion, although variations in the methacrylate conversion produced the most significant differences. Related to this issue, filler particle induction of inflammatory cytokines and chemokines in cells was demonstrated (Ansteinsson et al., 2009). Another study used a micromanipulation technique to examine various representative fillers used in dental composites. Either spherical or irregular filler particles as well as nanoclustered agglomerates were subjected to compressive testing, which provided force-displacement curves on both individual filler particles and nanoclusters. Differences in fracture behavior were observed, with the nanocluster filler producing multiple fracture events (Curtis et al., 2009a). The same group also found enhanced damage tolerance in cyclic fatigue loading of Filtek Supreme, hypothesized to be associated with its nanocluster filler morphology (Curtis et al., 2009b). Ultimately, a model of filler effects on composite properties was developed (Tanimoto et al., 2006).
The field of composite dental restoratives continues to propose and achieve significant and exciting advances in resin formulation, filler loading and modification, and curing methodologies and mechanisms. While most of the advances discussed herein remain in the research stage, the future both in regards to research and in clinical practice remains bright with exciting new developments translated into practice at an ever-increasing rate. With hundreds of millions of restorations performed each year, continuing research into practical advances and successful clinical implementation of composite restoratives are both critical to oral care, aesthetics, and functional restoration.
Footnotes
Acknowledgements
The authors gratefully acknowledge NSF Grant 0626023, and NIH/NIDCR Grants DE10959 and DE018233. This work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Dental and Craniofacial Research or the National Institutes of Health.