
Abstract
Amelogenesis imperfecta (AI) can be either isolated or part of a larger syndrome. Junctional epidermolysis bullosa (JEB) is a collection of autosomal-recessive disorders featuring AI associated with skin fragility and other symptoms. JEB is a recessive syndrome usually caused by mutations in both alleles of COL17A1, LAMA3, LAMB3, or LAMC2. In rare cases, heterozygous carriers in JEB kindreds display enamel malformations in the absence of skin fragility (isolated AI). We recruited two kindreds with autosomal-dominant amelogenesis imperfecta (ADAI) characterized by generalized severe enamel hypoplasia with deep linear grooves and pits. Whole-exome sequencing of both probands identified novel heterozygous mutations in the last exon of LAMB3 that likely truncated the protein. The mutations perfectly segregated with the enamel defects in both families. In Family 1, an 8-bp deletion (c.3446_3453del GACTGGAG) shifted the reading frame (p.Gly 1149Glufs*8). In Family 2, a single nucleotide substitution (c.C3431A) generated an in-frame translation termination codon (p.Ser1144*). We conclude that enamel formation is particularly sensitive to defects in hemidesmosome/basement-membrane complexes and that syndromic and non-syndromic forms of AI can be etiologically related.
Introduction
Amelogenesis imperfecta (AI), by the strict definition, refers to inherited malformations of dental enamel that occur in the absence of non-dental phenotypes (Witkop, 1989). Non-syndromic forms of autosomal-dominant AI can be caused by mutations in ENAM (Hu and Yamakoshi, 2003) and FAM83H (Kim et al., 2008). ENAM is a tooth-specific gene (Meredith et al., 2009), and the phenotype caused by ENAM mutations is limited to dental enamel (SG Simmer et al., 2012). Sometimes the phenotype is mild when a single ENAM allele is mutated and is manifested by well-delineated enamel pits, whereas little or no enamel forms when both alleles are affected (Ozdemir et al., 2005). FAM83H mutations cause autosomal-dominant hypocalcified amelogenesis imperfecta (ADHCAI). Patients with disease-causing FAM83H mutations have one wild-type allele and one allele with a truncation mutation that induces a dominant-negative pathogenic mechanism (Kweon et al., 2013). Unlike enamelin, which is secreted, FAM83H is intracellular and expressed in tissues other than developing teeth. The tooth-specificity of the phenotype may relate to ameloblasts (the cells that make enamel) being the cell-type that is most severely affected by pathology resulting from a truncated FAM83H protein.
Amelogenesis imperfecta also describes the enamel phenotype in syndromes. Junctional epidermolysis bullosa (JEB) is a collection of recessive inherited disorders featuring skin fragility and AI caused by defects in genes encoding the components of hemidesmosome/basement-membrane complexes (Masunaga, 2006). JEB cases with well-described enamel defects have been reported for COL17A1 (McGrath et al., 1996; Pasmooij et al., 2007), ITGB4 (Pulkkinen et al., 1998), and genes encoding laminin-332, such as LAMB3 (Buchroithner et al., 2004). The enamel is characterized by deep pits and grooves that might increase the risk for dental caries (Wright et al., 1994). A single defective allele for a gene that causes JEB (when both alleles are defective) may be manifested as autosomal-dominant amelogenesis imperfecta (ADAI) with little or no apparent skin fragility. The first reported cases were two heterozygous daughters of a COL17A1 compound heterozygous (g.3514ins25/p.G627V) proband with JEB. Both daughters inherited the same defective COL17A1 allele (p.G627V) and showed extensive enamel defects with no signs of skin fragility (McGrath et al., 1996). In the next generation, a single copy of the COL17A1 p.G627V allele was passed on to the proband’s granddaughter, who showed enamel defects and mild skin fragility, being the first reported case of autosomal-dominant JEB (Almaani et al., 2009). The COL17A1 p.G627V substitution lies within the collagen triple-helical domain and causes abnormal folding of the COL17A1 trimer, making it susceptible to degradation (Tasanen et al., 2000). In another case, a COL17A1 p.T239fs*52 mutation that caused ADAI in the heterozygous condition reduced the amount of wild-type COL17A1, since the mutant mRNA was subjected to nonsense-mediated decay and degraded (Murrell et al., 2007).
ADAI in heterozygous carriers of JEB have also been reported for LAMA3 and LAMB3 mutations. A JEB proband was a LAMA3 compound heterozygote (p.G163Dfs*30/p.A1495V), while his mother and brother carried only a single defective LAMA3 allele (p.G163Dfs*30) and had ADAI manifested as pitted enamel defects (Yuen et al., 2012). More recently, a p.E1133Gfs*27 mutation in a single LAMB3 allele segregated perfectly with the enamel phenotype in a four-generation ADAI family (Poulter et al., 2013). In this case, the defect was in the last exon, so the mRNA transcript likely escaped nonsense-mediated decay and allowed for translation of the truncated protein. Here, we report two novel LAMB3 mutations causing ADAI, confirming that heterozygous defects in LAMB3 can cause ADAI and providing new insight into the types of mutations that cause dominant, non-syndromic AI.
Materials & Methods
Patients
Two unrelated families, of Turkish and Iranian descent, with irregular hypoplastic ADAI phenotypes were selected from the AI families we have recruited for genetic studies. Clinical examinations were performed and blood samples were collected with the understanding and written consent of each participant according to the Declaration of Helsinki. The study protocol was independently reviewed and approved by the Institutional Review Boards at Seoul National University Dental Hospital, the University of Istanbul, and the University of Michigan.
Whole-exome Sequencing
Whole-exome sequencing was performed with genomic DNA isolated from the probands of the two AI families to identify the disease-causing mutation(s). Exomes were captured with Illumina TruSeq DNA sample prep kit, and 90-bp paired-end sequencing data were obtained with Illumina HiSeq 2000 (Macrogen, Seoul, Korea; Theragen Bio Institute, Suwon, Korea). Alignment of the sequencing data to the NCBI human reference genome (NCBI build 37.2, hg19) was performed, and the sequence variations were annotated with the dbSNP build 135.
Polymerase Chain-reaction and DNA Sequence Analyses
Primer pairs for the last exon of the LAMB3 gene (sense, 5′-CTGGAGAGGCATGAAGCTG; antisense, 5′-GCTGCAGC TCAGGGTAATCT) were designed with Primer 3 on the Web (http://frodo.wi.mit.edu/primer3/). PCR amplifications were performed with the HiPi DNA polymerase premix (Elpis Biotech, Taejeon, Korea) and purified with a PCR Purification Kit (Elpis Biotech). DNA sequencing was performed at the DNA sequencing center (Macrogen, Seoul, Korea).
Results
Clinical Findings
The proband of Family 1 was a 6.5-year-old girl presenting with sensitive teeth. Her mother, who was the source of information concerning the pedigree and affection status of unexamined individuals, reported that the biological father had dental problems similar to those of the proband (Fig. 1A). The mother’s dentition was within normal limits (data not shown). The proband’s enamel for both the primary and secondary dentition was thin (hypoplastic), with deep grooves and pitting defects (Fig. 1B). Radiographic examination revealed severe generalized enamel hypoplasia. The enamel was generally more radiopaque than dentin, but not in all places. Taurodontism was evident in both the primary and secondary molars (Fig. 1D).
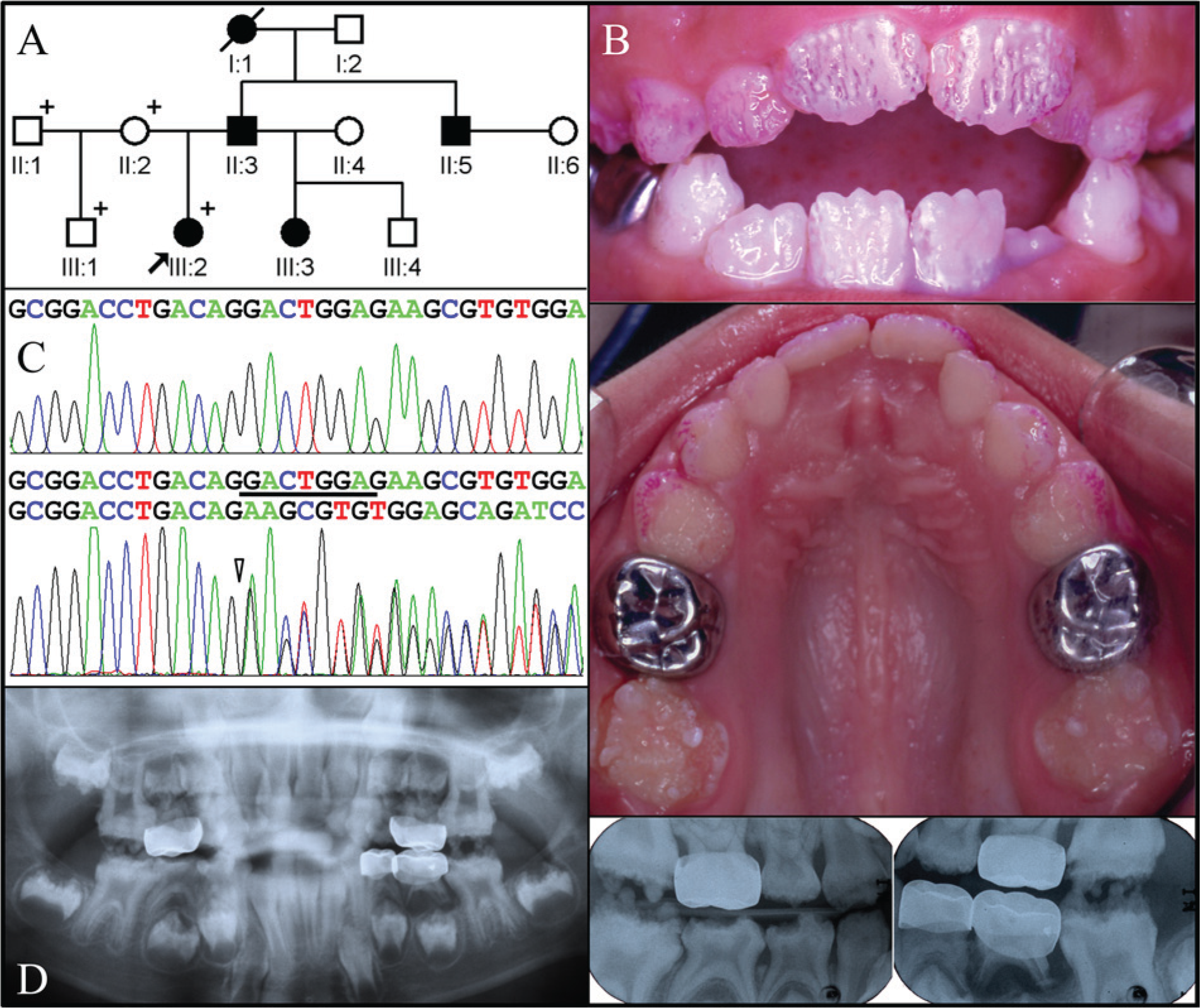
Clinical and mutational analysis of Family 1. (
The proband of Family 2 also presented with enamel hypoplasia in all teeth, a trait she inherited from her mother (Fig. 2). The proband complained of thermal hypersensitivity and displayed a distinctive pattern of enamel defects characterized by deep grooves and pits that led us to consider that the enamel phenotype in these two families might be caused by defects in a common gene. In both probands, the linear grooves in the incisors tended to run vertically, with the most pronounced grooves extending from between the mamelons and delineating regions where differentiating ameloblasts derived from adjacent growth centers met during tooth formation. In both families, the pattern of inheritance was autosomal-dominant, and neither family reported a medical history of skin fragility. Differences between the phenotypes were minor: The Family 2 proband displayed hypertrophic gingiva in the maxillary anterior, and her radiographs showed little or no taurodontism.
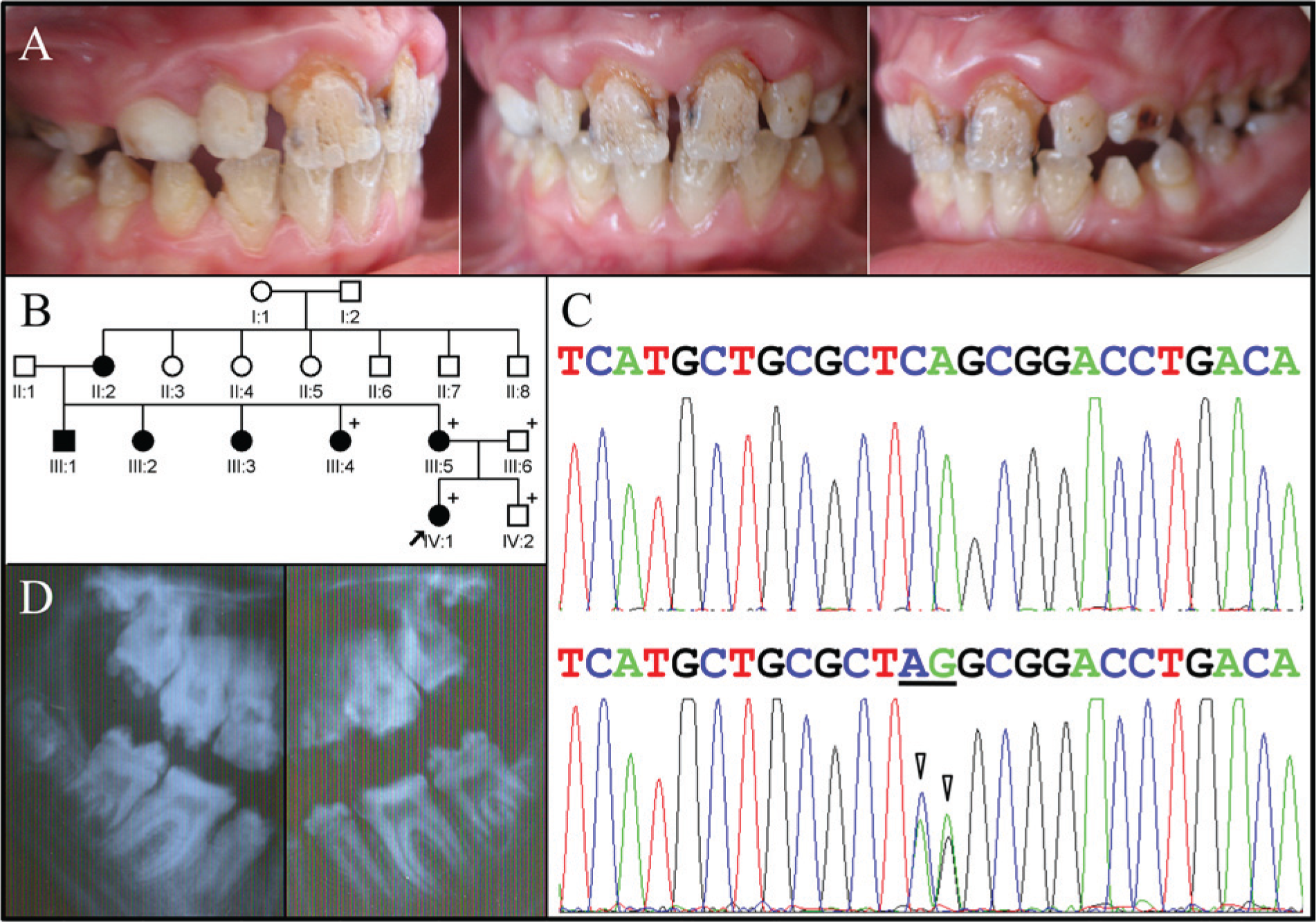
Clinical and mutational analysis of Family 2. (
Mutation Results
We obtained potential disease-causing sequence variations from the whole-exome sequence data by combining the annotation files of the two probands and searching for genes with potentially deleterious sequence variations in both probands. Heterozygous, destructive sequence variations in LAMB3 were ascertained that are not listed in the 1000 Genomes Project. The LAMB3 mutation identified in Family 1 (Fig. 1C) was an 8-bp deletion in the last exon (c.3446_3453delGACTGGAG) that shifted the reading frame and caused premature translation termination following the addition of 7 extraneous amino acids (p.Gly1149Glufs*8). The proband of Family 2 had a LAMB3 nonsense mutation (c.C3431A, p.Ser1144*) in the last exon (Fig. 2C).
For clarity, the cDNA designation for the disease-causing mutation in Family 2 requires explanation. The three affected members of Family 2 (III:4, III:5, IV:1) differed from the LAMB3 cDNA reference sequence (NM_000228.2) at two adjacent positions (c.C3431A and c.A3432G). The two unaffected members (III:6, IV:2) showed the reference sequence at both positions. Therefore, we considered the designation c.3431_3432delinsAG. However, the G at c.3432 is listed as a single nucleotide polymorphism (SNP; rs1049607), so the disease-causing mutation was likely a single nucleotide change in LAMB3 that preceded this SNP and resulted in a nonsense mutation and should be designated c.C3431A.
Distribution of the putative disease-causing mutations among the recruited members of both families was analyzed by Sanger sequencing of exon 23 PCR amplification products and demonstrated complete segregation of the LAMB3 mutations with the disease phenotype. There are now 3 known LAMB3 mutations associated with ADAI, all causing premature translation termination in the last coding exon (Fig. 3A). An alignment of the exon 23-encoded protein sequence at the LAMB3 C-terminus shows strong conservation of sequence length and 5 perfectly conserved amino acids between fish and humans that are lost in the mutant LAMB3 alleles (Fig. 3B).

Analysis of ADAI-causing LAMB3 mutations. (
Discussion
Laminin-332 (formerly laminin V) is a component of basement membranes and comprised of 3 subunits. Laminin is anchored to epithelial cells by collagen XVII and can activate integrin signaling through α6/β4 receptors. The C-terminal regions of LAMA, LAMB1, and LAMB2 and other possible variants form the long arm of Laminin V, which contains sites for receptor-mediated cell attachment and promotion of neurite outgrowth (Beck et al., 1990). Defects in both alleles of LAMA3, LAMB3, LAMC2, and COL17A1 cause JEB, while defects in both alleles of ITGA6 and ITGB4 cause JEB with pyloric atresia. All of these conditions are recessive and associated with enamel malformations. With the 2 ADAI mutations reported here, there are now 3 single-allele LAMB3 mutations associated with ADAI in the absence of other signs of JEB. All 3 mutations prematurely terminate translation in the last coding exon. The wild-type LAMB3 protein has 1172 amino acids. The truncated forms have 1131, 1143, and 1148 amino acids of normal LAMB3 sequence, so the sizes of the C-terminal deletions are only 41, 29, and 24 amino acids, respectively. Since the defect in Family 2 is a simple nonsense mutation, it appears that the absence of the normal C-terminal amino acids from the LAMB3 protein is sufficient to cause the enamel phenotype, even without the short (7- and 28-amino-acid) extraneous sequences translated downstream from the reading frame shifts. Truncation mutations in one allele, however, are not sufficient to cause skin fragility or the other non-dental phenotypes associated with JEB. But why should enamel formation be so sensitive to defects in hemidesmosome/basement-membrane complexes?
Although some reports suggest that laminin is expressed by secretory ameloblasts, it has long been known that the basement membrane of the inner enamel epithelia fenestrates and disappears under differentiating ameloblasts (Reith, 1967). Laminin epitopes are lost as pre-ameloblasts initiate amelogenin secretion (Inai et al., 1991) and do not appear again until ameloblasts transition to the maturation stage (Nanci et al., 1993). In place of the basement membrane under secretory-stage ameloblasts is a mineralization front apparatus that initiates and elongates thin ribbons of amorphous calcium phosphate (Beniash et al., 2009; JP Simmer et al., 2012). Secretory-stage ameloblasts are not strongly attached to the extracellular matrix, and migrate past each other to give the enamel layer a decussating pattern of enamel rods.
Lama3 null mice do not show defects in the inner enamel epithelium, which are attached by hemidesmosomes to a Laminin-332 basement membrane. Dental abnormalities in Lama3 null mice first appear during ameloblast differentiation, downstream of adhesion, when the basement membrane is being degraded and replaced by the mineralization front (Ryan et al., 1999). Part of the reason for the downstream effects is the dysregulation of enamel protein genes. Laminin-332 activates integrin signaling primarily via α6β4 receptors (Chiang et al., 2011). Amelogenin (Amelx) and enamelin (Enam) are specifically and strongly up-regulated in the enamel organs of Itgb6 null mice (Mohazab et al., 2013), suggesting that degradation of the hemidesmosome/basement-membrane complexes de-represses enamel protein expression, synchronizing replacement of the basement membrane complex by the mineralization front apparatus and presumably establishing new matrix-cell interactions. Ameloblast cell pathology also occurs in Enam null and Ambn (ameloblastin) defective mice, both of which fail to form the enamel mineralization front (Fukumoto et al., 2004; Hu et al., 2008).
The onset of enamel formation involves fenestration of the basement membrane, extension of ameloblast processes into irregularities on the pre-dentin surface, expression of enamel proteins, and the initial formation of enamel ribbons. The nearly synchronous ameloblast cell pathology in Lama3, Itgb6, Enam, and Ambn defective mice suggests that coordinated replacement of hemidesmosome/basement-membrane complexes with the mineralization front apparatus is critical for ameloblast physiology and presumably places special demands upon these complexes beyond that normally required for cell adhesion. This likely explains why single-allele defects in COL17A1, LAMA3, LAMB3, and potentially other genes involved in the etiology of JEB can be manifested by severe enamel malformations, while both alleles of these genes must be mutated to induce the skin fragility and other manifestations in JEB patients.
Our Family 1 is one (AI#23) of the first AI families we recruited for genetic studies (Kim et al., 2006). With this report, 14 of the AI-causing mutations have been identified in these 24 families. Among the 11 discernible autosomal-dominant cases, we identified 1 LAMB3, 3 ENAM, and 5 FAM83H disease-causing mutations. The proven candidate genes for ADAI should be expanded to include FAM83H, ENAM, LAMA3, LAMB3, and COL17A1. X-linked AI is caused by defects in AMELX. The current list of proven candidate genes for non-syndromic autosomal-recessive AI includes MMP20, KLK4, WDR72, C4orf26, and SLC24A4. There are also many genes, such as CNNM4 and FAM20A, that have been proven to cause recessive syndromes where AI can be the predominant phenotype at the time of initial diagnosis. The complexity of the potential genetic etiology in a patient presenting with inherited enamel defects makes mutational analyses of target genes based upon genotype-phenotype correlations largely impractical. Whole-exome analyses are currently the best way to discover the disease-causing mutation in patients with AI (Wang et al., 2013).
Non-syndromic AI-causing mutations have now been found in genes encoding secreted enamel matrix proteins and proteases (AMELX, ENAM, C4orf26, MMP20, KLK4), intracellular (FAM83H, WDR72), transmembrane (SLC24A4, COL17A1), and basement membrane (LAMA3, LAMB3) proteins. These genes, however, account for only about half of all cases, so more causative genes await discovery. Suspected candidate genes include AMBN, AMTN, ODAM, LAMC2, ITGA6, ITGB4, and ITGB6.
Footnotes
This work was supported by grants from the Bio & Medical Technology Development Program (2011-0027790), the Science Research Center grant to Bone Metabolism Research Center (2012-0000487), the Korea Research Foundation Grant funded by the Korean Government (MEST), and the
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.