
Abstract
Periodontitis is a chronic infectious disease driven by dysbiosis, an imbalance between commensal bacteria and the host organism. Periodontitis is a leading cause of tooth loss in adults and occurs in about 50% of the US population. In addition to the clinical challenges associated with treating periodontitis, the progression and chronic nature of this disease seriously affect human health. Emerging evidence suggests that periodontitis is associated with mechanisms beyond bacteria-induced protein and tissue degradation. Here, we hypothesize that bacteria are able to induce epigenetic modifications in oral epithelial cells mediated by histone modifications. In this study, we found that dysbiosis in vivo led to epigenetic modifications, including acetylation of histones and downregulation of DNA methyltransferase 1. In addition, in vitro exposure of oral epithelial cells to lipopolysaccharides resulted in histone modifications, activation of transcriptional coactivators, such as p300/CBP, and accumulation of nuclear factor–κB (NF-κB). Given that oral epithelial cells are the first line of defense for the periodontium against bacteria, we also evaluated whether activation of pathogen recognition receptors induced histone modifications. We found that activation of the Toll-like receptors 1, 2, and 4 and the nucleotide-binding oligomerization domain protein 1 induced histone acetylation in oral epithelial cells. Our findings corroborate the emerging concept that epigenetic modifications play a role in the development of periodontitis.
Introduction
The oral mucosa maintains a functional barrier that protects against chemical, physical, and biological insults, including bacterial lipopolysaccharides (LPS) (Elias 1983; Elias and Choi 2005). Chronic inflammation and/or abrupt microbial imbalance (dysbiosis) dynamically alter cellular and molecular events, leading to the development of periodontal disease, inflammatory bowel disease, irritable bowel syndrome, celiac disease, and colitis, among other diseases (Baumgart and Carding 2007; Kornman 2008; De Palma et al. 2010; Jiao et al. 2014). In the oral cavity, dysbiosis evolves from the overgrowth of microbial commensals, or “pathobionts,” and results in oral epithelial injury, bone loss, and periodontitis, which is a common disease affecting more than 50% of the adult population (Hugoson and Norderyd 2008). This overgrowth of pathobionts drives activation of specific pathogen recognition receptors (PRRs), including NOD1 and Toll-like receptors (TLRs). In addition to activating PRRs, evidence suggests that bacteria induces extensive epigenetic modifications, activates proinflammatory pathways, and increases virulence (Casadesus and Low 2006; Zhang et al. 2013; Barros and Offenbacher 2014). Although methylation of the DNA adenine has been associated with bacterial virulence, the impact of other epigenetic modifications on periodontal disease, particularly acetylation of histones, remains unclear (Larsson et al. 2015).
Two major types of epigenetic modifications are DNA methylation and histone acetylation. Interestingly, targeted modulation of the immune response modifies DNA (cytosine-5-)–methyltransferase 1 and histone deacetylases (HDACs), 2 key controllers of DNA methylation and histone modifications (Kim et al. 2013). In fact, acetylation of histones relaxes the chromatin conformation and leads to enhanced transcription of inflammatory genes, including the p300/CBP histone acetyltransferase target gene NF-κB, and several proinflammatory cytokines, commonly upregulated in periodontitis. Although the impact of histone modifications in periodontal diseases is unclear, NF-κB signaling appears to play a key role in connecting histone modifications to disease progression. NF-κB activates innate immunity and protects against infections (Dev et al. 2011). However, chronic activation of NF-κB signaling causes osteoclast differentiation and bone resorption (reviewed in Abu-Amer 2013). Similar to periodontal disease, other inflammatory conditions, such as rheumatoid arthritis, are sensitive to deregulated NF-κB (Sánchez-Pernaute et al. 2008). In both cases, compromised equilibrium between activation of innate immunity and decreased NF-κB activity leads to undesirable tissue destruction even after pathogens have been eradicated. Altogether, we hypothesize that pathobiont-induced periodontal disease mediates acetylation of the chromatin of oral epithelial cells.
In this study, we explored the intrinsic ability of bacterial commensals to prime NF-κB activity through epigenetic modifications. We found that common pathogens of the oral cavity, including Porphyromonas gingivalis and Fusobacterium nucleatum, induce epigenetic modifications such as acetylation of histones and downregulation of DNA methyltransferase 1 (DNMT1), particularly in epithelial cells. These modifications lead to enhanced activation of p300/CBP transcriptional coactivators and increased NF-κB activity. Furthermore, PRRs, including NOD1 and TLR1/2 and TLR4, induce histone modifications. Collectively, our findings suggest the involvement of histone modifications in the process of pathobiont-induced periodontal disease.
Materials and Methods
See the Appendix for an extended description of the experimental approaches.
Animal Model for Periodontal Disease
This study was executed in concordance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines for preclinical studies. The mouse studies were approved by the University of Michigan Committee on Use and Care of Animals. Specific pathogen-free wild-type mice aged 11 to 12 wk received sterile surgical silk sutures around the first and second maxillary molars and maintained for 10 d to induce periodontitis. After euthanasia, formalin-fixed maxillae were subjected to micro–computed tomography (micro-CT) image analysis. To assess alveolar bone loss, we measured the distance between the cementoenamel junction and alveolar bone crest (CEJ-ABC) at distobuccal sites for the first molars and mesiobuccal sites for the second molars in 3-dimensional images.
Cell Line, Immunohistochemistry, Immunofluorescence, Antibodies, Reagents, and Immunoblotting
Normal human oral keratinocyte (NOK-SI) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (10% fetal bovine serum [FBS], 1% antibiotics). Trichostatin A (TSA; Sigma-Aldrich, St. Louis, MO, USA) was used as positive control for histone acetylation. Immunohistochemistry and immunofluorescence were performed with antibodies for acetyl-histone H3 (Lys9; Cell Signaling Technology, Beverly, MA, USA), phospho-p65 (Ser536; Cell Signaling Technology), DNMT1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and p300/CBP (Cell Signaling Technology), followed by isothiocyanate (FITC) or tetramethylrhodamine isothiocyanate (TRITC)–conjugated secondary antibodies and Hoechst 33342. For western blot, whole-cell lysate was loaded into sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinyl difluoride membrane (Immobilon; Millipore, Billerica, MA, USA), incubated with DNMT1 (Santa Cruz Biotechnology) antibody, and visualized using ECL SuperSignal (Pierce Biotechnology, Waltham, MA, USA).
LPS and PRR Agonists
F. nucleatum (ATCC 25586) was cultured on Trypticase soy agar with 5% blood plates under anaerobic incubation (80% N2; 10% H2; 10% CO2) and further inoculated into Trypticase soy broth supplemented with hemin (5 µg/mL) and vitamin K (0.5 µg/mL) to obtain F. nucleatum liquid culture. Bacterial liquid cultures were inactivated by heating at 98°C for 10 min and then used for an in vitro assay. The source and working concentrations of TLR1/2, TLR4, NOD1 agonists, and LPS were PAM3CSK4 (1 µg/µL; Bachem, Bubendorf, Switzerland), Lipid A (5 µg/µL; Sigma-Aldrich), KF1B (1 µg/mL; gift from Dr. Koichi Fukase, Osaka University), Escherichia coli 055:B5 LPS (1 µg/mL; Sigma-Aldrich), and LPS preparation of P. gingivalis W83 and 33277 (1 µg/µL; extracted from Pg) (Darveau and Hancock 1983; Padial-Molina et al. 2013).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Statistical analysis of the NF-κB p65, DNMT1, p300/CBP, and ac.H3 stains was performed by 1-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison tests. Statistical analysis of bone loss was performed by using an unpaired 2-tailed Student’s t test (parametric). Asterisks denote statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, and P > 0.05 [NS]).
Results
The Ligature-Induced Periodontal Model Displays Increased Inflammatory Cell Infiltrate, Epithelial Recession, and Loss of Periodontal Ligament and Alveolar Bone
The formation of a biofilm provides support for growth of indigenous bacteria that play a role in periodontal disease and its complications (Demmer and Desvarieux 2006; Signat et al. 2011). In this study, we used an in vivo ligature-induced periodontitis model that recapitulates the host-bacteria interactions in periodontal disease progression, which is characterized by intense inflammatory infiltrate and bone loss (Graves et al. 2012; Abe and Hajishengallis 2013; Jiao et al. 2013). Ten days after placing silk ligatures on the first and second molars, mice had severe loss of alveolar bone, as shown on the micro-CT images (Fig. 1A, arrowhead) and further confirmed in histological sections (Fig. 1B, C, arrowhead). Littermate control mice without ligatures retained normal bone crests (Fig. 1D–F, arrows), indicating that periodontal disease was induced by the accumulation of bacterial commensals and silk ligature. Mice with ligatures had high levels of inflammatory cell infiltrate in the gingival epithelium (Fig. 1C, arrowhead). These mice also had reduced periodontal ligaments fibers (PDLs) (Fig. 1C) compared to control mice with well-organized PDLs and healthy gingival papillae and gingival epithelium attached to the cement-gingival junction (CEJ) (Fig. 1F). Severe bone loss was statistically significant between ligature and control animal groups as judged by the distance between the cementoenamel junction and alveolar bone crest (CEJ-ABC) (*P < 0.05) (Fig. 1G).
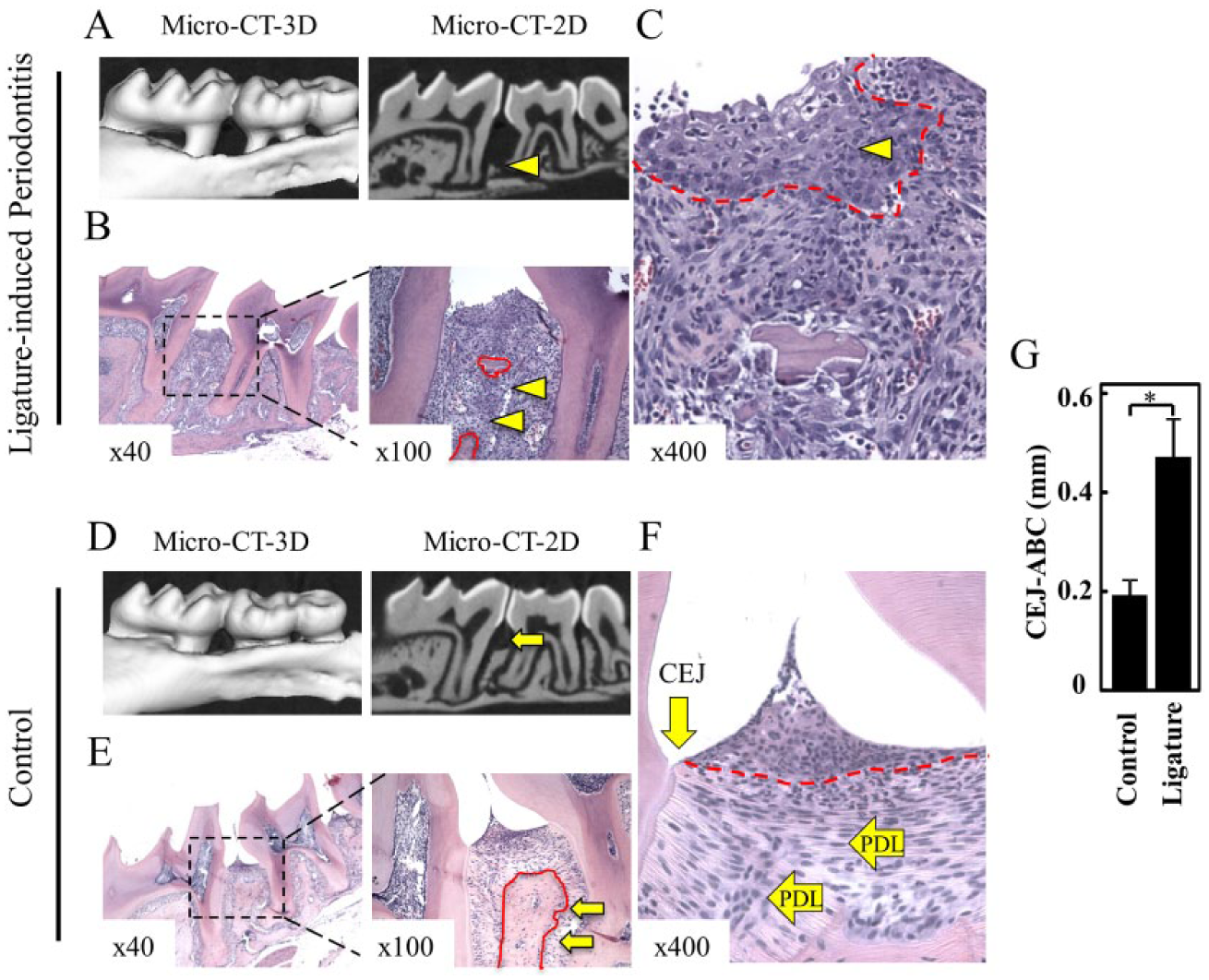
Ligature-induced model for periodontal disease. (
LPS-Induced Downregulation of DNA Methyltransferase 1
Epigenetic modifications are associated with structural changes in chromatin, which are independent from changes in the DNA sequence. While acetylation of histones interferes with chromatin rearrangement and gene transcription, DNA methylation is commonly associated with gene silencing. Therefore, epigenetic control of gene expression is achieved by balancing histone modifications and DNA methylation, which is regulated by DNMT1 (Jenuwein and Allis 2001). We examined whether bacterial commensals influence the levels of DNMT1 in periodontal disease. We found that loss of DNMT1 expression was decreased in the affected gingival epithelium of mice with periodontal disease compared to controls (Fig. 2A, ***P < 0.001). Similar to murine oral epithelial cells, human gingival keratinocytes had reduced nuclear DNMT1 in response to LPS stimuli (P. gingivalis–W83) (Fig. 2B, ***P < 0.001). Western blot data confirmed that DNMT1 was reduced after 15 min of LPS treatment (Fig. 2C). Similar to our results, LPS also reduces DNMT1 levels in skin cells (de Camargo Pereira et al. 2013), suggesting a common molecular mechanism among cells of epithelial origin.
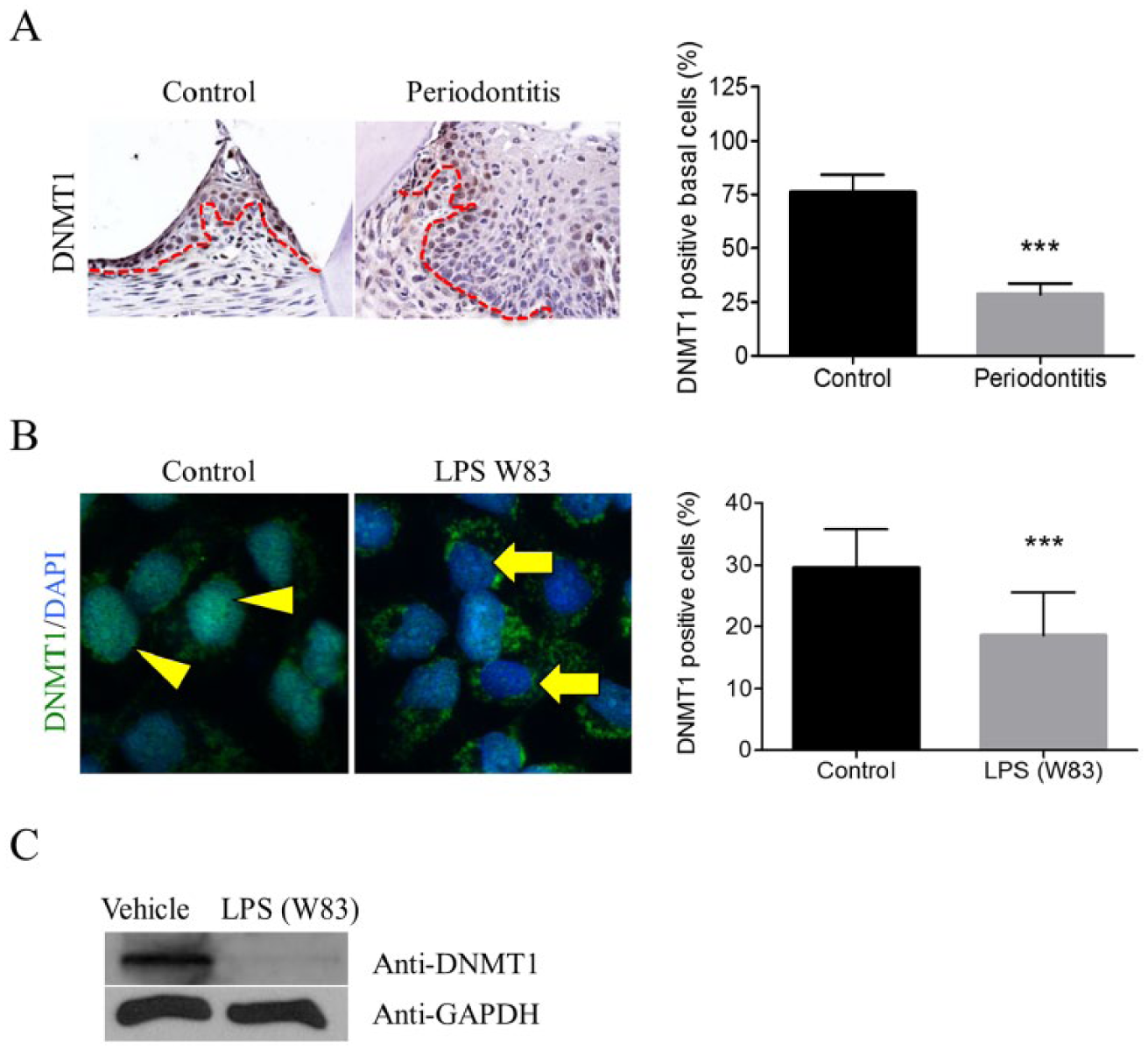
Lipopolysaccharide (LPS) downregulates DNA methyltransferase 1 (DNMT1). (
Dysbiosis and LPS-Induced Histone Modifications in Oral Epithelial Cells
Evidence suggests a potential association between dysbiosis and methylation of PRRs (Benakanakere et al. 2015). However, the effects of dysbiosis-induced acetylation of histones remain unknown. We found that oral epithelial cells from ligature-induced periodontitis mice had increased acetylation of histone H3 (Fig. 3A, B, ***P < 0.001), suggesting that acquisition of a dysbiotic condition is sufficient to induce histone modifications. However, it is unknown whether histone acetylation is due to a dysbiotic condition or is the result of the presence of multiple environmental factors during periodontal disease progression. To better characterize this process, we exposed human oral epithelial cells to the products of different bacterial strains, including LPS from P. gingivalis, E. coli, and heat-inactivated F. nucleatum, and analyzed histone H3. Interestingly, LPS from P. gingivalis (LPS W83 and LPS 33277, *P < 0.05) and heated-inactivated F. nucleatum (*P < 0.05) induced acetylation of histone H3 quickly (15 min) compared to LPS from E. coli (6 h, ***P < 0.001) (Fig. 3C, D). We used Trichostatin A (TSA), a histone deacetylase inhibitor, as a positive control for H3 acetylation. As expected, TSA induced global chromatin decondensation and acetylation of histones (Fig. 3C, D, ***P < 0.001).
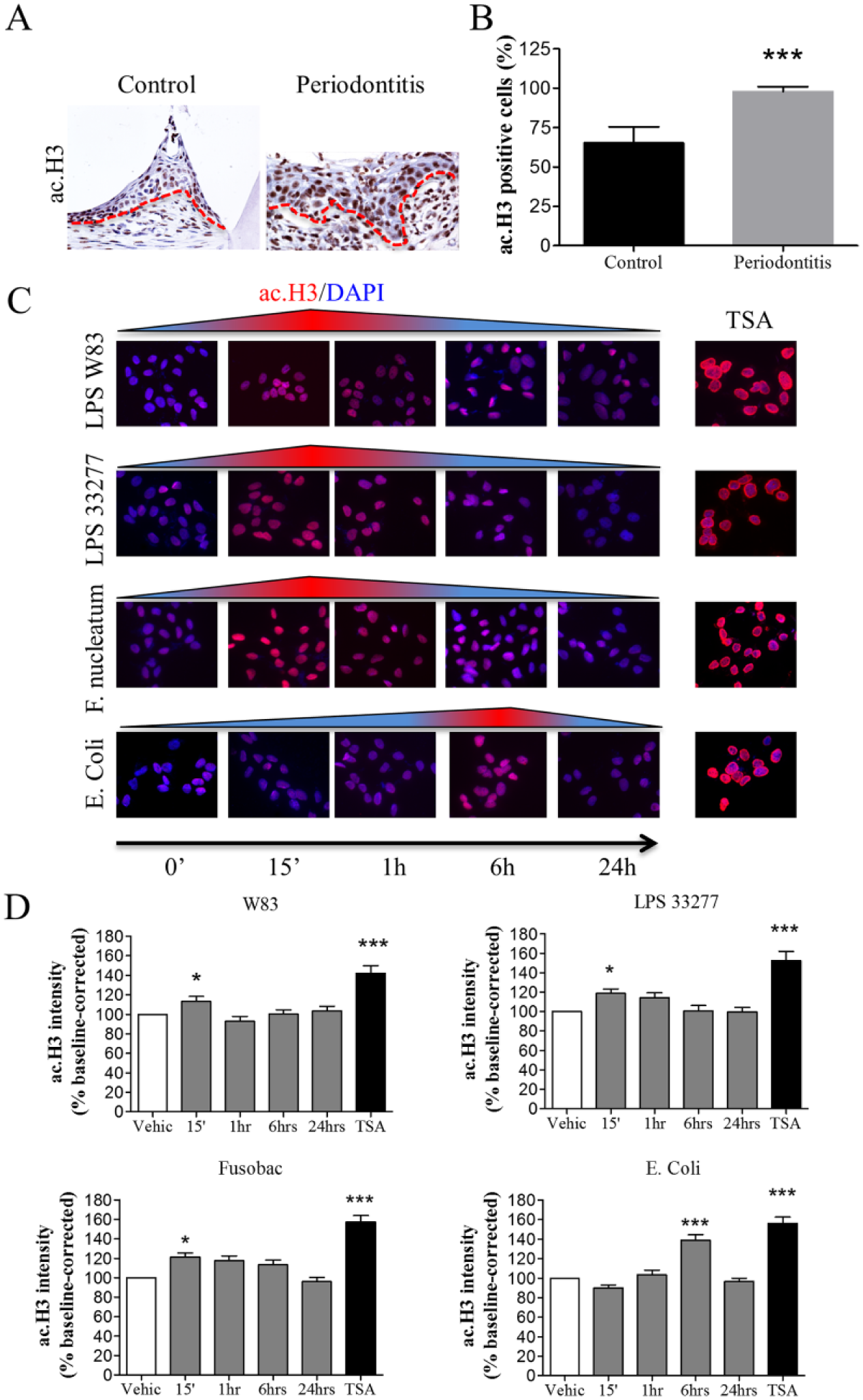
Pathobionts and lipopolysaccharide (LPS) induce acetylation of histones in murine periodontitis and in human oral epithelial cells. (
LPS-Treated Oral Epithelial Cells Show Activation of the Transcriptional Coactivator p300/CBP and Nuclear Translocation of NF-κB
Acetylation of histones by LPS suggests a potential role for p300/CBP in periodontal disease. P300/CBP is involved in many transcriptional mechanisms and coordinates diverse signals, including cellular differentiation, proliferation, and apoptosis, and regulates transcription via chromatin acetylation (reviewed in Brownell and Allis 1996). Activation of p300/CBP also results in transcriptional stimulation of several proinflammatory cytokines, including interleukin (IL)–1, IL-2, IL-8, and IL-12, which are commonly upregulated in periodontitis. Indeed, we found that LPS exposure resulted in the accumulation of p300/CBP in oral epithelial cells (Fig. 4A, B, *P < 0.05) and nuclear accumulation of NF-κB (Fig. 4C). Interestingly, activation and accumulation of NF-κB occurred within 1 min of LPS exposure and lasted for 15 min (Fig. 4C, ***P < 0.001). Collectively, our findings suggest that accumulation of pathobionts adjacent to oral epithelial cells results in robust modification of histones and accumulation of p300/CBP and NF-κB.
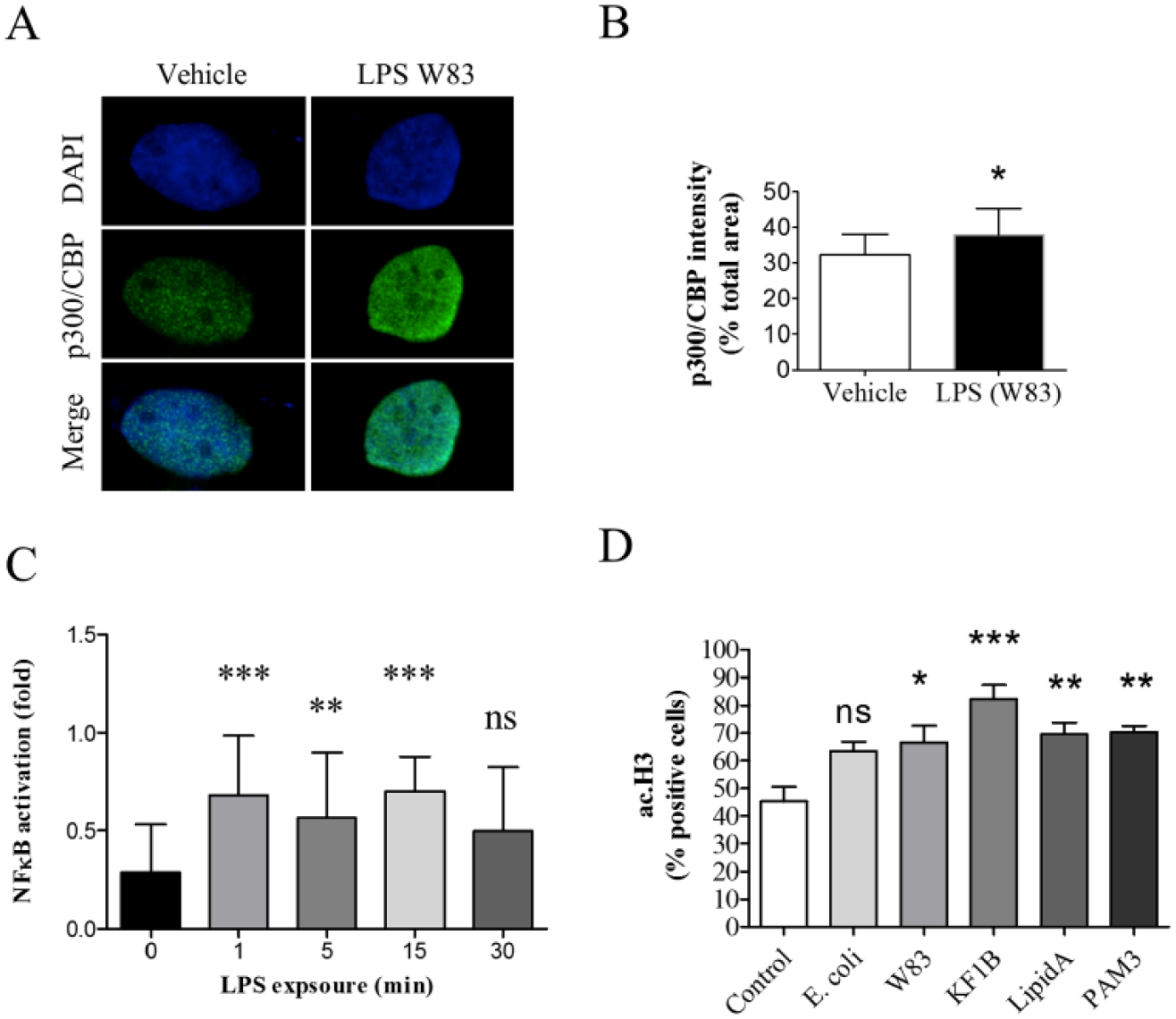
Lipopolysaccharide (LPS) family of pathogen recognition receptor (PRRs; TLRs and NOD1) activates p300/CBP and nuclear factor (NF)–κB. (
PRR-Induced Acetylation of Histones
PRRs, which mediate the detection of pathogens in the oral cavity, comprise membrane-bound and cytoplasmatic molecules (Inohara et al. 2001). TLRs are membrane-bound PRRs, while NOD1 is a cytoplasmatic receptor–like molecule that recognizes bacterial products in the cytosol of host cells. In addition, TLRs and NOD1 are associated with the development and progression of periodontitis (Mahanonda and Pichyangkul 2007; Jiao et al. 2013). We examined whether acetylation of histones in periodontal disease is mediated by PRRs. We activated NOD1 using the synthetic ligand KF1B (Masumoto et al. 2006), TLR1/2 heterodimers using the agonist PAM3 (Funderburg et al. 2011), and TLR4 using the agonist LipidA (Mata-Haro et al. 2007). Notably, the activated PRRs induced acetylation of histone H3 in human oral epithelial cells (Fig. 4D). Interestingly, activation of NOD1 resulted in the strongest acetylation of histone H3 (***P < 0.001) compared to activation of TLRs 1, 2, and 4 (**P < 0.01). In addition, direct activation of PRRs resulted in higher H3 acetylation compared to activation by LPS, suggesting that PRRs support higher stimulatory signals from pathobionts (W83, *P < 0.05). Interestingly, E. coli–derived LPS failed to induce significant acetylation of histone H3 after 1 h (NS, P > 0.05).
Discussion
Epigenetic modifications are gaining substantial attention as an alternative regulator of gene expression. Although epigenetic modifications encompass a large number of epigenetic processes (e.g., noncoding RNAs, DNA and histone methylations, and histone acetylation, among others), only a handful of these processes have been studied in periodontal disease (Zhang et al. 2010; Benakanakere et al. 2015; reviewed in Larsson et al. 2015). DNA methylation is the epigenetic process most studied in periodontal disease, but few studies have examined histone modifications, particularly following exposure to bacterial LPS. Larsson et al. (2012) showed that LPS stimulates H3 methylation and acetylation of H3 and H4 in B cells, while others observed that HDAC inhibitors, which induce histone acetylation, have a protective effect on LPS-induced bone resorption (Cantley et al. 2011). Some of the most unexpected findings about histone modifications came from studies demonstrating reactivation of human immunodeficiency virus 1 (HIV-1) and Epstein-Barr latent virus by LPS-driven histone modifications (Imai et al. 2009; Imai et al. 2012). We found that P. gingivalis LPS and heat-inactivated F. nucleatum induce abrupt but short-lived acetylation of histone H3 (Lys9) in oral epithelial cells, a process associated with the activation and accumulation of p300/CBP. Interestingly, E. coli LPS resulted in a delayed but powerful induction of histone acetylation compared to P. gingivalis and F. nucleatum. Similar to our findings, E. coli LPS have been shown to elicit a robust and broad response in vivo compared to P. gingivalis LPS (Liu, Desta, et al. 2008). The presence of E. coli LPS has also been previously shown to potently induce vascular cell adhesion protein 1 (VCAM-1) and tumor necrosis factor α (TNF-α) at local and distant sites compared to P. gingivalis LPS-restricted local induction of VCAM-1 (Liu, Desta, et al. 2008). The use of P. gingivalis extracts also shows variation in inducing T-cell responses among different strains (reviewed in Ellis and Kuehn 2010). Also, the clinical impact of distinct forms of P. gingivalis LPS has been recently demonstrated to differentially affect bone loss and the expression of interleukins (Marchesan et al. 2012). The use of whole bacteria lysate versus purified LPS is also of importance. LPS isolated from bacteria is considered the most potent immune-stimulating component of the outer membrane vesicles from gram-negative bacteria. The use of isolated LPS is known to trigger proinflammatory response, leading to septic shock upon high concentrations (Opal 2007). However, chemical extraction of LPS results in the isolation of LPS from its outer membrane vesicles, which are composed of lipoproteins and outer membrane proteins. Therefore, the use of outer membrane vesicles present in whole bacteria produces a different and likely more pathogen-specific immunostimulation of the host compared to LPS alone (Lee et al. 2008). Besides differences in LPS potency, our results may elude to other epigenetic modifications particularly driven by P. gingivalis. In a recent report, P. gingivalis LPS was shown to hypermethylate TLR2, resulting in reduced host innate defense and enhanced disease susceptibility (Benakanakere et al. 2015). Such P. gingivalis immunosuppressant advantage may have a direct impact on histone acetylation of epithelial cells and in the activation of transcription genes. In fact, we have shown that administration of the TLR1/2 heterodimers agonist resulted in enhanced histone acetylation. As such, hypermethylation of TLR2 by P. gingivalis LPS could affect the levels of histone acetylation compared to E. coli LPS. Although exciting, the molecular circuitry underlying LPS and histone modifications in periodontal disease remains unknown. As an immune-inflammatory infection, periodontal disease involves many PRRs, including the NOD1 and TLR families that are responsible for detecting pathobionts following dysbiosis in the oral cavity. This process is primarily initiated in epithelial cells from the oral mucosa, which serves as the first line of defense against pathogens. Given that PRRs are essential to initiating the innate immune response in epithelial cells, we examined the effect of PRRs on histone acetylation in comparison with E. coli and P. gingivalis LPS. We showed for the first time that LPS-induced acetylation of histones is mediated by PRRs. Interestingly, the effects of the agonists for TLRs 1/2, 4, and NOD1 on histone acetylation were greater than LPS stimulation. Much of these results may have to do with the activation efficiency of agonists to their receptors compared to LPS binding. Indeed, Sun et al. (2010) have shown that P. gingivalis LPS induced TLR2, whereas TLR4 required very high concentrations of LPS. Interestingly, however, exposure to inactivated P. gingivalis resulted in a powerful activation of TLR4, suggesting the presence of other virulence factors (Sun et al. 2010). Similar to P. gingivalis, LPS from E. coli failed to activate TLR2, whereas whole E. coli was able to activate TLR2. These findings corroborate our findings with the use of PRR agonists, including Lipid A, KF1B, and PAM3, resulting in significant increased acetylation of histone 3 compared to LPS from E. coli and P. gingivalis.
These findings expand our knowledge about the molecular signaling mechanisms involved in periodontitis, particularly how dysbiosis epigenetically enhances cellular transcription. In addition, these findings suggest new pharmacological targets, such as histones and PRRs, for treating periodontal disease. Indeed, targeted therapy against PRRs has been used to treat Pseudomonas aeruginosa infection, which can cause lung, urinary tract, and kidney infections; generalized inflammation; and sepsis (Moalli et al. 2011). In addition to PRRs, we found that LPS also induced accumulation of p300/CBP. While the precise implications of p300/CBP in periodontal disease progression remain elusive, its control of NF-κB signaling is well documented and of interest to periodontal biology. The mammalian NF-κB family is a target gene of the transcriptional coactivator and histone acetyltransferase p300/CBP (Zhong et al. 2002). Activation of NF-κB signaling can be elicited through stimulation of cytokines and bacterial products; therefore, NF-κB appears to be a critical component of the innate immune response (Kim et al. 2006).
Gene expression is a well-controlled mechanism mediated by DNA methylation and histone modifications. These genetic regulations are not separate events; they are linked, fine-tuned, and provide cell-specific regulation of gene expression (Robertson and Wolffe 2000). While DNA methylation is associated with gene silencing, histone acetylation is associated with chromatin rearrangement and gene transcription. In epithelial cells, the presence of TLR2 promoter hypermethylation reduces cytokine signaling in a process rescued by the administration of DNMT inhibitors (Benakanakere et al. 2015). Our data demonstrated for the first time that LPS stimuli cause acetylation of oral epithelial cells (ac.H3-lys9) and reduced expression of DNMT1, suggesting active gene transcription. Together, the work from Benakanakere et al. (2015) and our findings demonstrate the dynamic between gene methylation and histone acetylation during dysbiosis. Similar to our findings, others found a decrease in DNMT1 and HDAC expression in gingival epithelial cells after stimulation with P. gingivalis and F. nucleatum (Yin and Chung 2011), as well as the demonstration of decreased DNMT1 expression following exposure to P. gingivalis LPS in keratinocytes (de Camargo Pereira et al. 2013). As a regulatory mechanism, active NF-κB binds to the DNMT1 promoter, resulting in increased DNMT1 protein expression (Liu, Liu, et al. 2008; Hong et al. 2013). Because NF-κB is a target gene of p300/CBP, there may be a molecular connection between p300/CBP and DNMT1 levels, but information regarding these potential interactions is limited. Furthermore, acetylation of the p300/CBP promoter at H3K9, reduced histone deacetylase activity, and recruitment of NF-κB result in the release of several cytokines (Barnes 2009).
Collectively, we have identified a role for PRRs in LPS-induced acetylation of histones in oral epithelial cells. Our findings highlight that the intricate process of epigenetic activation of transcription is a molecular balance between histone acetylation and DNMT1 activity and that the equilibrium between these epigenetic mechanisms leads to the accumulation of p300/CBP and NF-κB (Fig. 5). However, the precise regulatory mechanisms underlying the interactions between DNMT1, p300/CBP, and NF-κB remain poorly understood and require further characterization. It is also important to note that our in vitro data are derived from one immortalized cell line that contain unknown genetic alterations associated to its spontaneous immortalization process, and therefore NOK-SI cells may differ from normal oral epithelial cells.
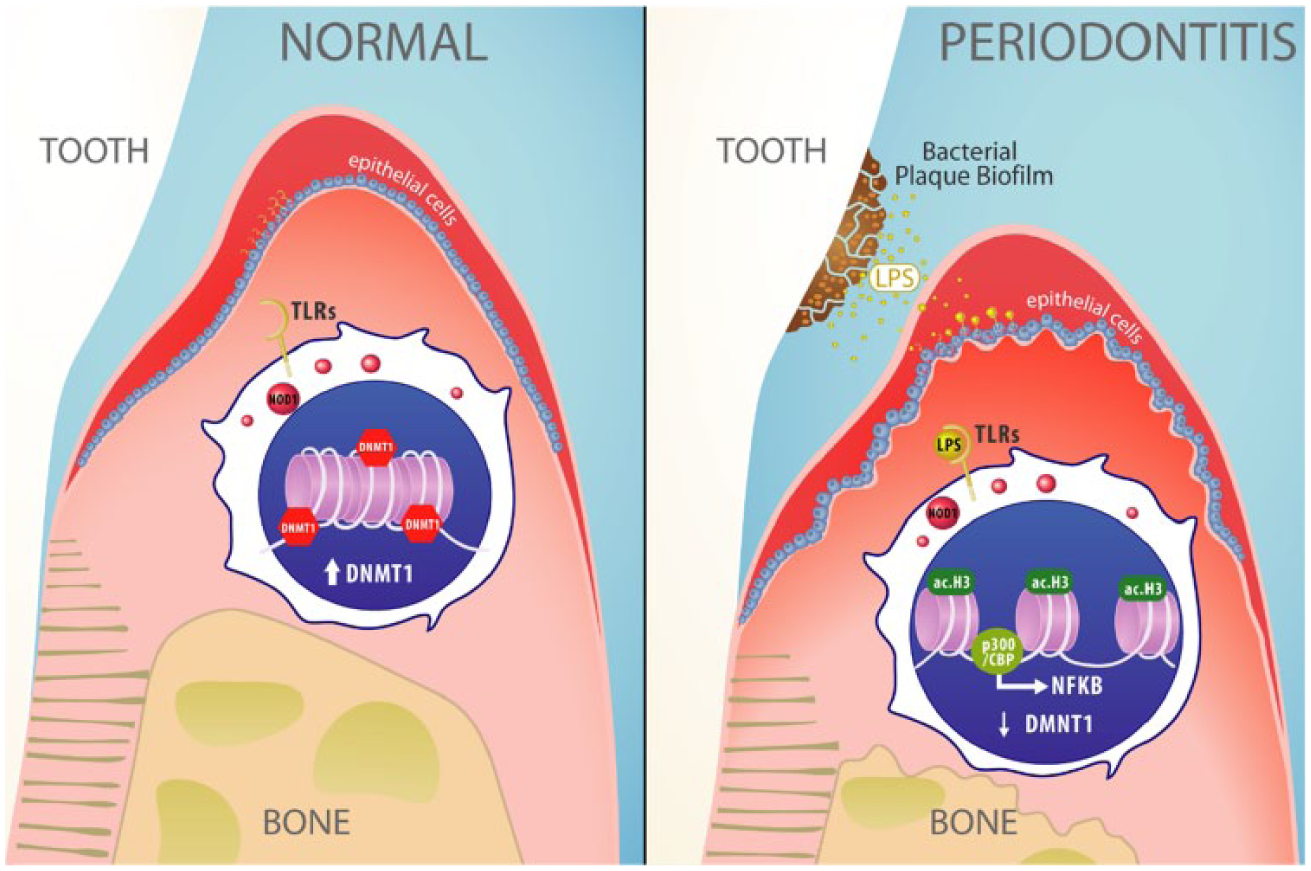
Proposed mechanism of epigenetic modulation of periodontal disease. Epithelial cells from the oral mucosa (blue) present nuclear DNA methyltransferase 1 (DNMT1) and low levels of acetylated histone H3. Upon dysbiosis, pathogen recognition receptors TLRs and NOD1 detect increased concentrations of bacterial lipopolysaccharide (LPS) and induce the acetylation of chromatin in epithelial cells. Acetylation of histone 3 (ac.H3) leads to chromatin remodeling, reduced levels of DNMT1, and recruitment of p300/CBP cotranscription factor, resulting in the activation of the immune response regulator nuclear factor (NF)–κB during dysbiosis.
Nonetheless, our results from murine and human tissues are complementary and suggest that disruption of dysbiosis-mediated epithelial injury may inhibit PRRs and that targeting the disruption of histone acetylation may provide the foundation for treating epigenetically relapsing periodontitis or for reestablishing tissue homeostasis.
Author Contributions
M.D. Martins, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript; Y. Jaio, L.O. Almeida, contributed to design and data acquisition, critically revised the manuscript; L. Larsson, contributed to conception, data acquisition, and analysis, critically revised the manuscript; C. Garaicoa-Pazmino, contributed to data analysis, critically revised the manuscript; J.M. Le, contributed to data acquisition, critically revised the manuscript; C.H. Squarize, R.M. Castilho, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; N. Inohara, contributed to conception, design, and data analysis, critically revised the manuscript; W.V. Giannobile, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
Acknowledgements
We thank Dr. Ricardo Teles for the gift of F. nucleatum and Dr. Fukase for the gift of KF-1B. We also thank the University of Michigan Medical School Host Microbiome Initiative for bacterial culture support.
This work was partially funded by CNPQ, CAPES (Coordination for the Improvement of Higher Education Personnel, process BEX 10714/13-8), and the University of Michigan School of Dentistry faculty grant.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.