
Abstract
The junctional epithelium (JE) is unique with regard to its wide intercellular spaces and sparsely developed intercellular junctions. Thus, knowledge of the molecular mechanisms that regulate the formation of the intercellular junctions of the junctional epithelium may be essential to understand the pathophysiology of the JE. HOK-16B cells, a normal human gingival epithelial cell line, were used to identify the molecules involved in the regulation of the formation of intercellular E-cadherin junctions between human gingival epithelial cells. Activation of c-Jun N-terminal kinase (JNK) disrupted the intercellular junctions through the dissociation of E-cadherin. The role of JNK in the formation of these E-cadherin junctions was further confirmed by demonstrating that JNK inhibition induced the formation of intercellular E-cadherin junctions. The upstream signaling of JNK was also examined. Activation of the small GTPase RhoA disrupted the formation of E-cadherin junctions between HOK-16B cells, which was accompanied by JNK activation. Disruption of these intercellular junctions upon RhoA activation was prevented when JNK activity was inhibited. In contrast, RhoA inactivation led to HOK-16B cell aggregation and the formation of intercellular junctions, even under conditions in which the cellular junctions were naturally disrupted by growth on a strongly adhesive surface. Furthermore, the JE of mouse molars had high JNK activity associated with low E-cadherin expression, which was reversed in the other gingival epithelia, including the sulcular epithelium. Interestingly, JNK activity was increased in cells grown on a solid surface, where cells showed higher RhoA activity than those grown on soft surfaces. Together, these results indicate that the decreased formation of intercellular E-cadherin junctions within the JE may be coupled to high JNK activity, which is activated by the upregulation of RhoA on solid tooth surfaces.
Introduction
The junctional epithelium (JE) forms a seal between tooth and the lamina propria of the gingiva, functioning as a barrier against the attack of microorganisms or toxic substances (Shimono et al. 2003; Bosshardt and Lang 2005). Its intercellular junctions are sparsely developed and its intercellular spaces are wide compared with those of other regions of the gingival epithelium (GE), which are tightly packed and have well-developed intercellular junctions. Thus, the JE allows the migration of protective leukocytes into the gingival sulcus (Schroeder and Listgarten 2003). However, bacterial toxins or bacteria may invade via these wide intercellular gaps in the JE when its protective balance is disrupted (Damek-Poprawa et al. 2011; Damek-Poprawa et al. 2013). Therefore, understanding the mechanisms that regulate the structural integrity of the JE is essential for preventing and treating gingivitis and periodontitis through maintaining or stabilizing this protective barrier.
The limited development of the intercellular junctions of the JE suggests that the mechanism regulating these junctions is altered. In this study, the molecular features associated with the development of intercellular E-cadherin junctions were analyzed to determine what factors lead to the restricted development of intercellular junctions of gingival epithelial cells. E-cadherin is critical to maintaining the structural integrity of the oral epithelium, including the JE (Ye et al. 2000; Bosshardt and Lang 2005). E-cadherin is also localized at the desmosome of the epithelium (Wheelock and Jensen 1992; Lewis et al. 1994; Lewis et al. 1997; Norvell and Green 1998) and is regarded as a crucial molecular component in the organization of the desmosomal complex (Wheelock and Jensen 1992; Lewis et al. 1994; Lewis et al. 1997; Norvell and Green 1998). For example, the E-cadherin–plakoglobin complex is required for desmosome organization (Presland and Dale 2000).
Recently, c-JUN NH3-terminal kinase (JNK) was shown to be involved in negatively regulating the formation of intercellular E-cadherin junctions (Lee et al. 2009; Lee et al. 2011). Active JNK phosphorylates β-catenin to prevent E-cadherin complexes from forming. In the present study, human gingival epithelial cells were analyzed to determine whether the formation of their intercellular E-cadherin junctions is dependent on JNK activity and which molecular control mechanisms regulate this activity. Understanding the molecular mechanisms controlling the formation of the intercellular junctions of oral epithelia may provide insight into the altered development of these junctions and suggest strategies defending against bacterial diseases in the oral cavity.
Materials and Methods
Reagents
The following antibodies for Western blotting were purchased from Cell Signaling Technology (Danvers, MA, USA): rabbit anti-JNK, rabbit anti–phospho-JNK (Thr183/Tyr185), rabbit anti–E-cadherin, rabbit anti-GAPDH, and horseradish peroxidase (HRP)–linked anti–rabbit IgG. Anti–phospho-myosin light chain (p-MLC) was purchased from Millipore (Darmstadt, Germany). Rabbit anti-ZO1 was purchased from ThermoFisher (Waltham, MA, USA). Rabbit anti–E-cadherin and rabbit anti–phospho-JNK for immunocytochemistry studies were also obtained from Cell Signaling Technology. Cy3-conjugated AffiniPure goat anti–rabbit IgG (H+L) (Cy3-IgG) was purchased from Jackson Immunoresearch Laboratories (West Grove, PA, USA). Antiserum against odontogenic ameloblast-associated protein (ODAM) (Nishio et al. 2010) was a gift from Dr. Joo-Cheol Park (Seoul National University, Seoul Korea). SP600125 (JNK inhibitor), SB203580 (p38 inhibitor), U0126 (ERK inhibitor), fluorescein isothiocyanate–labeled phalloidin (phalloidin-FITC), and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Exoenzyme C3 transferase from Clostridium botulinum (C3; RhoA inhibitor) and CNF-1 (RhoA activator) were obtained from Cytoskeleton (Denver, CO, USA). Col-Tgel was purchased from 101Bio (Palo Alto, CA, USA).
Preparation of Culture Dish Surfaces
Two different concentrations of fibronectin (Sigma-Aldrich) were used to coat hydrophobic petri dishes to allow for HOK-16B cell growth as a typical confluent epithelial layer (0.1 µg/cm2 fibronectin) or as a scattered cell layer (1 µg/cm2 fibronectin) at 37°C for 2 h. Laminin (Engelbreth-Holm-Swarm murine sarcoma basement membrane, Sigma-Aldrich; 1 µg/cm2) was also coated onto the dishes for the scattered cell growth experiments. Dishes were then washed with phosphate-buffered saline (PBS), blocked with 5% bovine serum albumin (BSA) for 1 h at 37°C, washed again with PBS, and kept at 4°C until use. Collagen gel surfaces were prepared by pouring a mixture of Col-Tgel component A and component B (20:1 v/v) onto culture dishes and incubating at 37°C for 45 min. Solidified gel surfaces were coated with 1 µg/cm2 of fibronectin for 1 h. Dishes were then washed with PBS.
Cell Culture
HOK-16B cells immortalized from periodontally healthy human retromolar gingival tissue were a gift from Dr. N.-H. Park (University of California, Los Angeles, USA) (Park et al. 1991). The cells were grown in KGM keratinocyte growth media supplemented with bovine pituitary extract, GA-1000 (gentamicin, amphotericin-B), hydrocortisone, rhEGF, insulin (recombinant human) (Lonza, Basel, Switzerland), and 1% penicillin. The HOK-16B cells established from keratizined gingival epithelium could be used for the study related with the JE, which is nonkeratinized. The regeneration of the JE from the keratinized gingival epithelium after a gingivectomy indicates that the nonkeratinized JE is derived from the keratinized gingival epithelium (Nishio et al. 2010). Furthermore, it is well known that the differentiation of epithelial cells depends on the substrates to which these cells attach (Mackenzie 1987; Pispa and Thesleff 2003; Jimenez-Rojo et al. 2012). In addition, we confirmed that HOK-16B cells expressed ODAM (Appendix Fig. 1), which is exclusively expressed in the JE, not in other oral epithelia, including sulcular epithelium and gingival epithelium (Park et al. 2007).
Transfections
pcDNA3-RhoA-Q63L (constitutively active RhoA: RhoA- CA), pcDNA3-RhoA-T19N (dominant-negative RhoA: RhoA- DN), pcDNA3-Flag-MKK7b2-JNK1a1 (constitutively active JNK1), pcDNA3-Flag-MKK7b2-JNK2a2 (constitutively active JNK2), and the lentiviral vector pCS-CG were purchased from Addgene (Cambridge, MA, USA). Lipofectamine LTX reagent and Plus reagent were purchased from Invitrogen (Carlsbad, CA, USA). Lentiviral vector M1.4, the virus packaging vector psPAX2, and the envelope protein vector pMD2G were gifts from Dr. Zang-Hee Lee (Seoul National University, Seoul Korea). M1.4-RhoA-CA, M1.4-RhoA-DN, pCS-CG-MKK7b2-JNK1a1, and pCS-CG-MKK7b2-JNK2a2 were generated by Cosmo Genetech (Seoul, Korea). The JNK1 shRNA (shJNK1) plasmid used for lentiviral transfections was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HEK293T cells (a gift from Dr. Z.-H. Lee) were transfected with recombinant vector, psPAX2, and pMD2G simultaneously using Lipofectamine LTX and Plus reagents. Lentiviruses generated by the transfected HEK293T cells were used to infect HOK-16B cells. Cells were selected by treating with puromycin.
Tissue Preparation and Immunohistochemistry
All experiments involving animals followed protocols approved by the Institutional Animal Care and Use Committee of Seoul National University (SNU-140828-1). Twelve-week-old C57BL/6 mice were euthanized by asphyxiation in carbon dioxide. Mandibular molars, including surrounding tissues, were excised and fixed with 4% phosphate-buffered paraformaldehyde. Tissues decalcified in a solution of 10% EDTA (pH 7.4) were embedded in paraffin. The deparaffinized sections, treated with a citrate buffer (pH 6.0) for antigen retrieval in a microwave oven, were incubated with primary antibodies against E-cadherin or p-JNK overnight at 4°C. These sections were then incubated with Cy3-IgG and examined using an LSM 700 confocal laser-scanning microscope (Zeiss, Oberkochen, Germany). Sections processed without primary antibody treatment were also examined to rule out any nonspecific staining.
Immunocytochemistry
HOK-16B cells were fixed with 4% w/v paraformaldehyde and permeabilized in 0.5% v/v Triton X-100. The cells were then incubated with primary antibodies against E-cadherin. Finally, cells were treated with Cy3-IgG. F-actin was stained with phalloidin-FITC. Images were acquired using an LSM 700 confocal laser-scanning microscope.
Statistical Analyses
The data represent the mean ± standard deviation of at least 3 samples. Statistical analyses were performed using a t test for normally distributed data or a signed rank sum test for nonnormally distributed data. P values less than 0.05 were considered significant.
Results
JNK Activity Is Critical to the Formation of Intercellular E-Cadherin Junctions between Human Gingival Epithelial Cells
HOK-16B cells formed a typical confluent epithelial layer and well-organized junctions expressing a high level of E-cadherin (Fig. 1A). We altered JNK activity to determine whether it was required for the formation of E-cadherin junctions between human gingival epithelial cells. The pharmacological activation of JNK with anisomycin and the constitutive activation of the MKK7b2-JNK1a1 fusion protein decreased E-cadherin expression at the cell junctions, leading to intercellular dissociations (Fig. 1A, B). Treatment of cells with the JNK inhibitor SP600125 prior to treatment with anisomycin eliminated the effect of anisomycin; pretreatment with SB203580, a p38 inhibitor, did not. These results indicate that JNK activity, not p38 activity, disrupts the formation of E-cadherin junctions (Fig. 1A). JNK regulation of E-cadherin was further confirmed by demonstrating that the intercellular junctions between HOK-16B cells could be restored by downregulating JNK activity, even in conditions where the cell junctions were naturally disrupted by growth on strongly adhesive surfaces. HOK-16B cells, which had grown scattered on dishes coated with large amounts of fibronectin or laminin (data not shown) and did not develop E-cadherin junctions, formed a typical confluent epithelial layer when JNK activity was inhibited by SP600125 or when the cells expressed shJNK1 (Fig. 1C, D). However, JNK did not regulate the expression level of E-cadherin under these experimental conditions (Appendix Fig. 2). Moreover, other types of mitogen-activated protein kinases (MAPKs), such as ERK and p38, were not associated with the formation of E-cadherin junctions (Appendix Fig. 3). These results indicate that JNK activity is specifically associated with the formation of the intercellular E-cadherin junctions between human gingival epithelial cells.
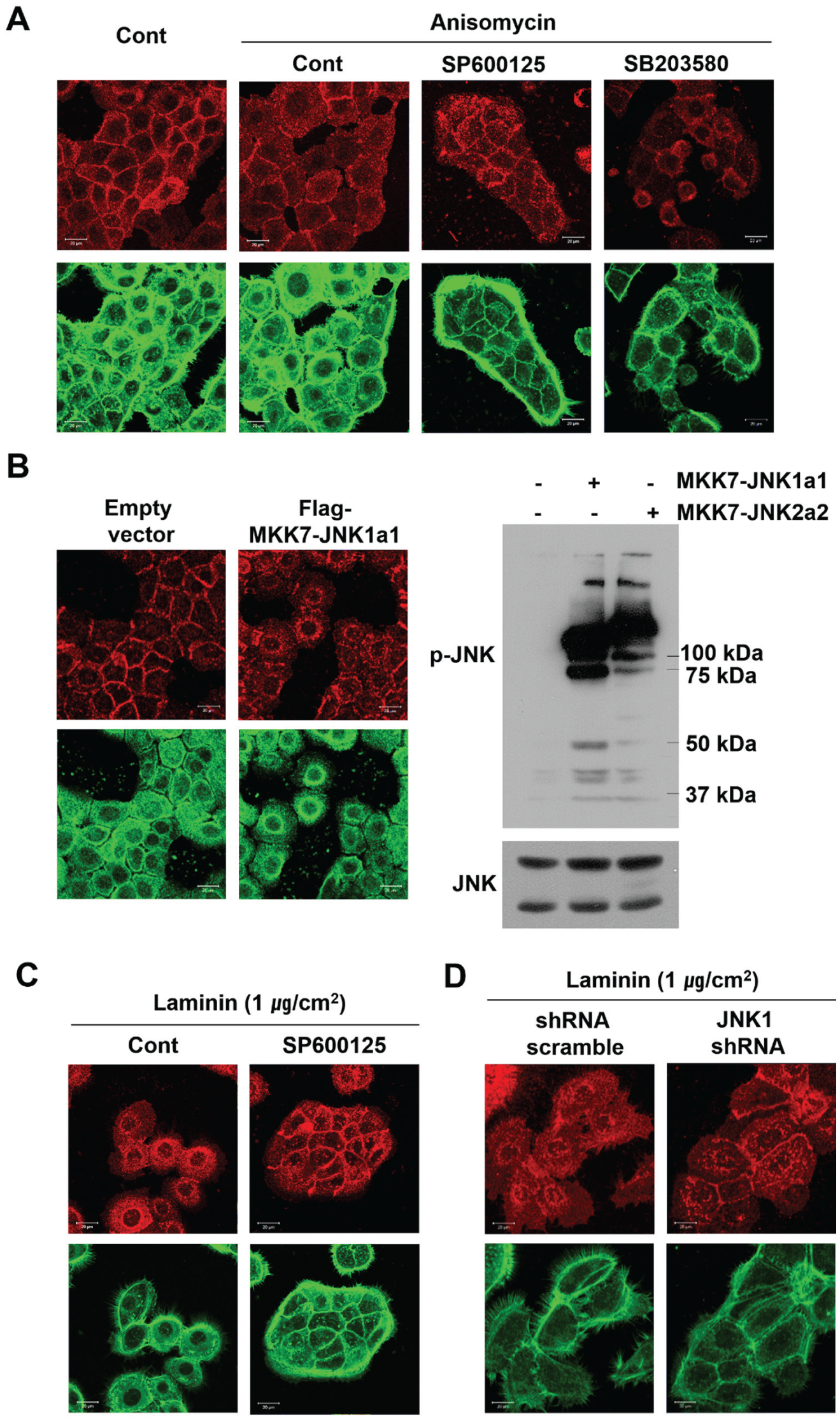
c-Jun N-terminal kinase (JNK) regulates the formation of intercellular E-cadherin junctions between HOK-16B cells in vitro. The formation of E-cadherin junctions was determined by examining the expression level of E-cadherin (upper panels). F-actin was stained with fluorescein isothiocyanate (FITC)–phalloidin (lower panels). (
RhoA Activity Disrupts the Intercellular Junctions between Human Gingival Epithelial Cells
RhoA controls the dynamics of actin filaments, which are one of the major components of E-cadherin junctions. It has also recently been reported to regulate various essential cell activities, such as proliferation and survival, in addition to cell migration and the formation of stress fibers (Ridley and Hall 1992; Yang et al. 2011; Yang and Kim 2012). Furthermore, RhoA is reportedly associated with the various functions of cell junctions. Rho GTPases remodel actin-based structures on endothelial cells to facilitate the migration of leukocytes through the junctions between endothelial cells. RhoA activation also increases the permeability of junctions between endothelial cells (Essler et al. 1998; Wojciak-Stothard et al. 1998). Based on these reports, we examined the role of RhoA in regulating the formation of E-cadherin junctions between human gingival epithelial cells. CNF-1, a specific RhoA activator, downregulated E-cadherin expression at the intercellular junctions, widening the gaps between neighboring cells (Fig. 2A). Constitutive activation of RhoA also dissociated the cell junctions (Fig. 2B). To confirm the function of RhoA as a negative regulator of intercellular junctions, HOK-16B cells growing in a scattered pattern were treated with C3, a specific RhoA inhibitor, in petri dishes coated with a large amount of fibronectin (a highly adhesive surface). Upon treatment with C3, the cells aggregated to form a confluent epithelial layer, accompanied by the formation of intercellular E-cadherin junctions (Fig. 2C). The dominant-negative inhibition of RhoA activity also induced cells to develop E-cadherin junctions, even when cultured on a highly adhesive surface (Fig. 2D). Taken together, these results indicate that RhoA is a negative regulator of intercellular E-cadherin junctions.
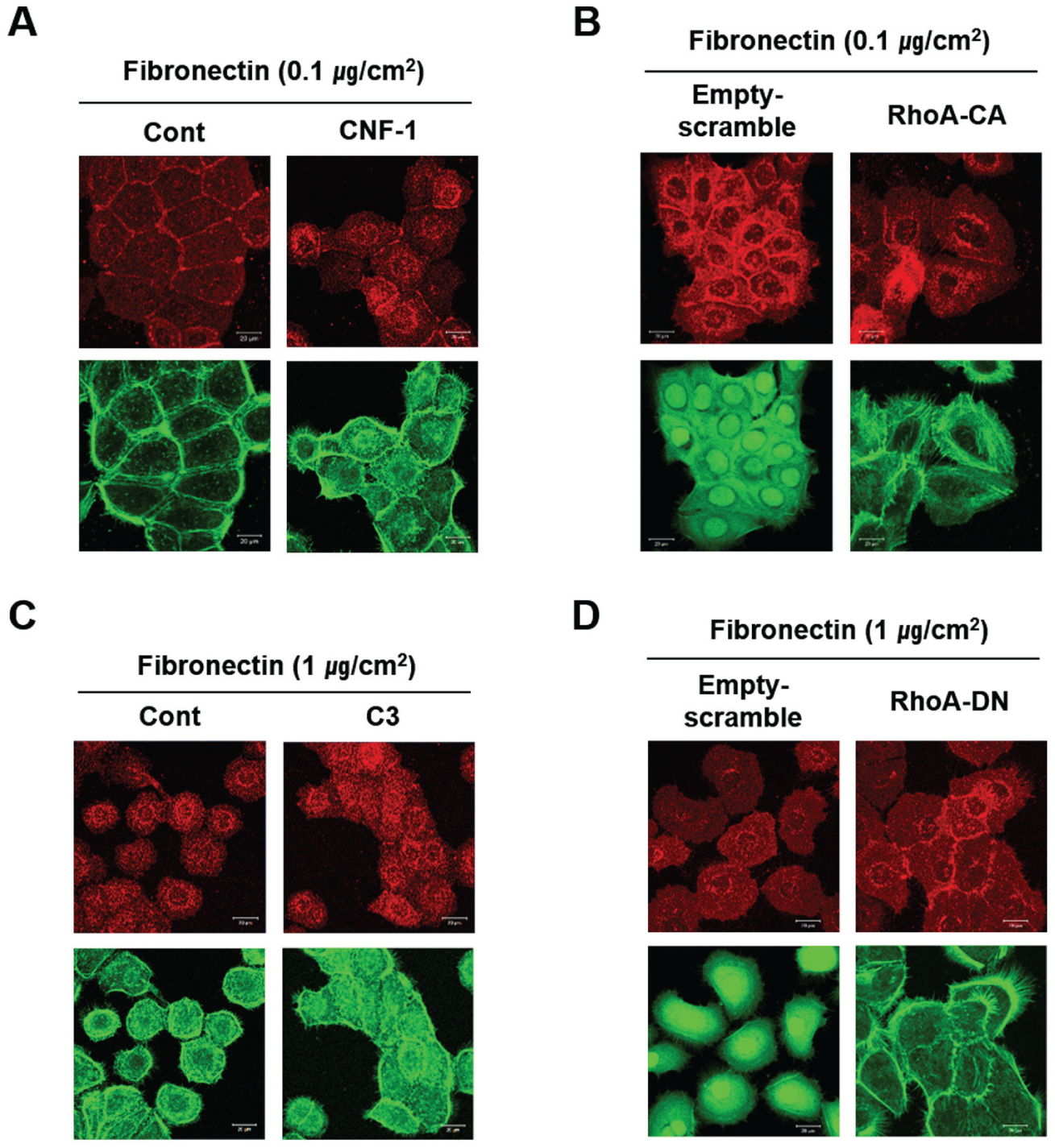
RhoA inhibits the formation of intercellular E-cadherin junctions between HOK-16B cells in vitro. The formation of E-cadherin junctions was determined by examining the expression level of E-cadherin (upper panel of each picture). F-actin was stained with fluorescein isothiocyanate (FITC)–phalloidin (lower panel of each picture). (
RhoA Disrupts the Intercellular E-Cadherin Junctions between Human Gingival Epithelial Cells by Activating JNK
RhoA activation induced by treating cells with CNF-1 or by transfecting the cells with a viral vector expressing RhoA-CA upregulated JNK phosphorylation (Fig. 3A, B). In contrast, RhoA inhibition induced by treating cells with C3 or by transfecting them with a viral vector expressing RhoA-DN downregulated JNK phosphorylation (Fig. 3C, D). Furthermore, disruption of the intercellular junctions resulting from RhoA activation was completely blocked when cells were pretreated with the JNK inhibitor SP600125 (Fig. 3E, F). These results suggest that JNK functions at a downstream point in the RhoA signaling pathway to disrupt the intercellular junctions between human gingival epithelial cells.
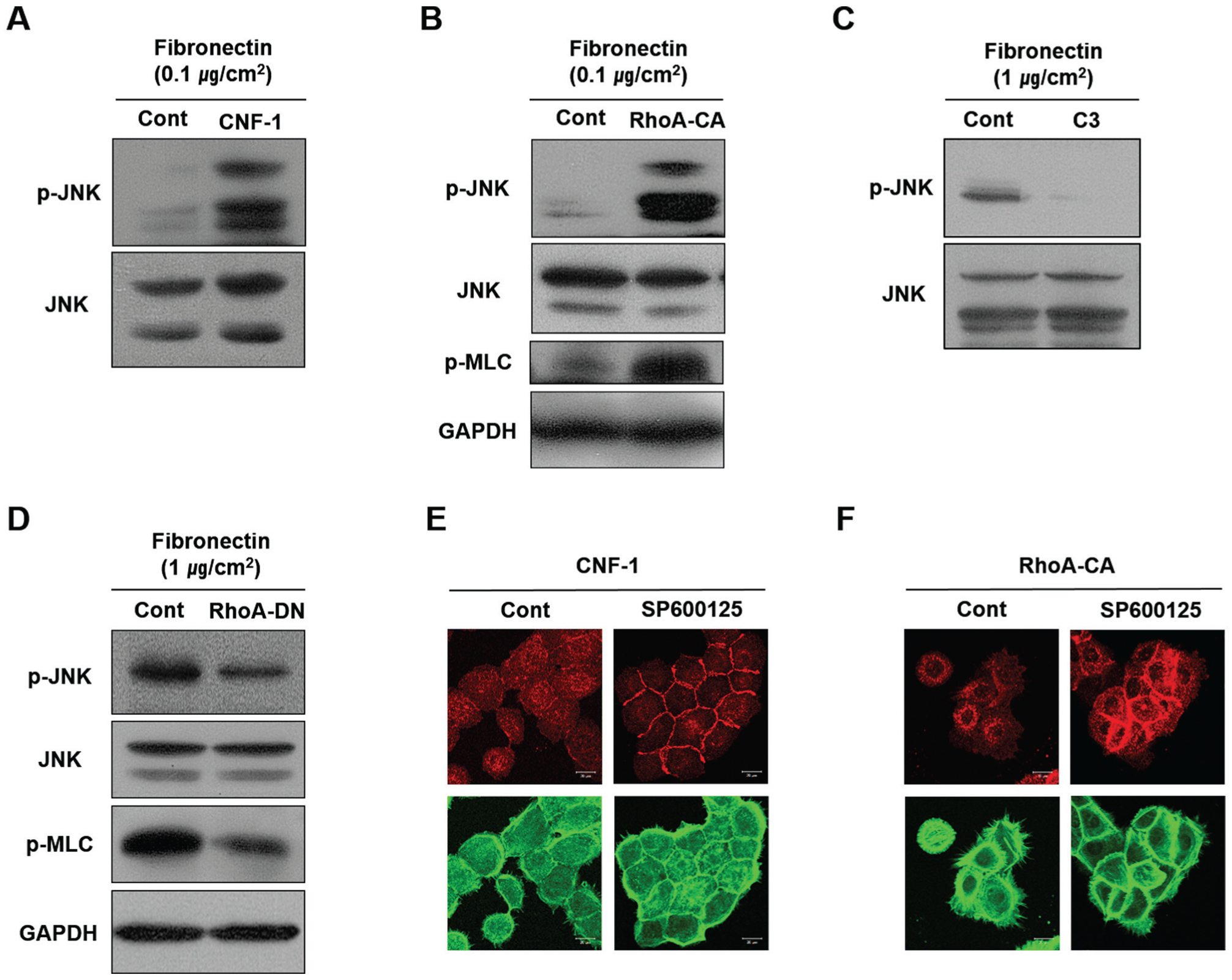
RhoA regulates the formation of intercellular E-cadherin junctions between HOK-16B cells by controlling JNK activity. (
JNK Is Highly Active in Junctional Epithelial Cells, but E-Cadherin Expression within the Intercellular Junctions of These Cells Is Low
The levels of p-JNK and E-cadherin expression in the GE of mouse molars were evaluated to confirm the existence of an inverse relationship between their expression levels. The JE of mouse molars was identified by the expression of ODAM (Nishio et al. 2010) and by its specific ultrastructural phenotype (Appendix Fig. 4). Immunohistochemical analysis of the JE showed that the level of E-cadherin in the intercellular junctions was substantially lower than that shown in other regions of the GE, including the sulcular epithelium (SE) (Fig. 4). The low expression of E-cadherin within the JE is common among different species. E-cadherin expression is reported to be lower in the JE in humans as well (Ye et al. 2000). As expected, expression levels of p-JNK were higher in the JE than in the SE (Fig. 4). These results suggest that higher JNK activity may be associated with the poor development of E-cadherin junctions within the JE.
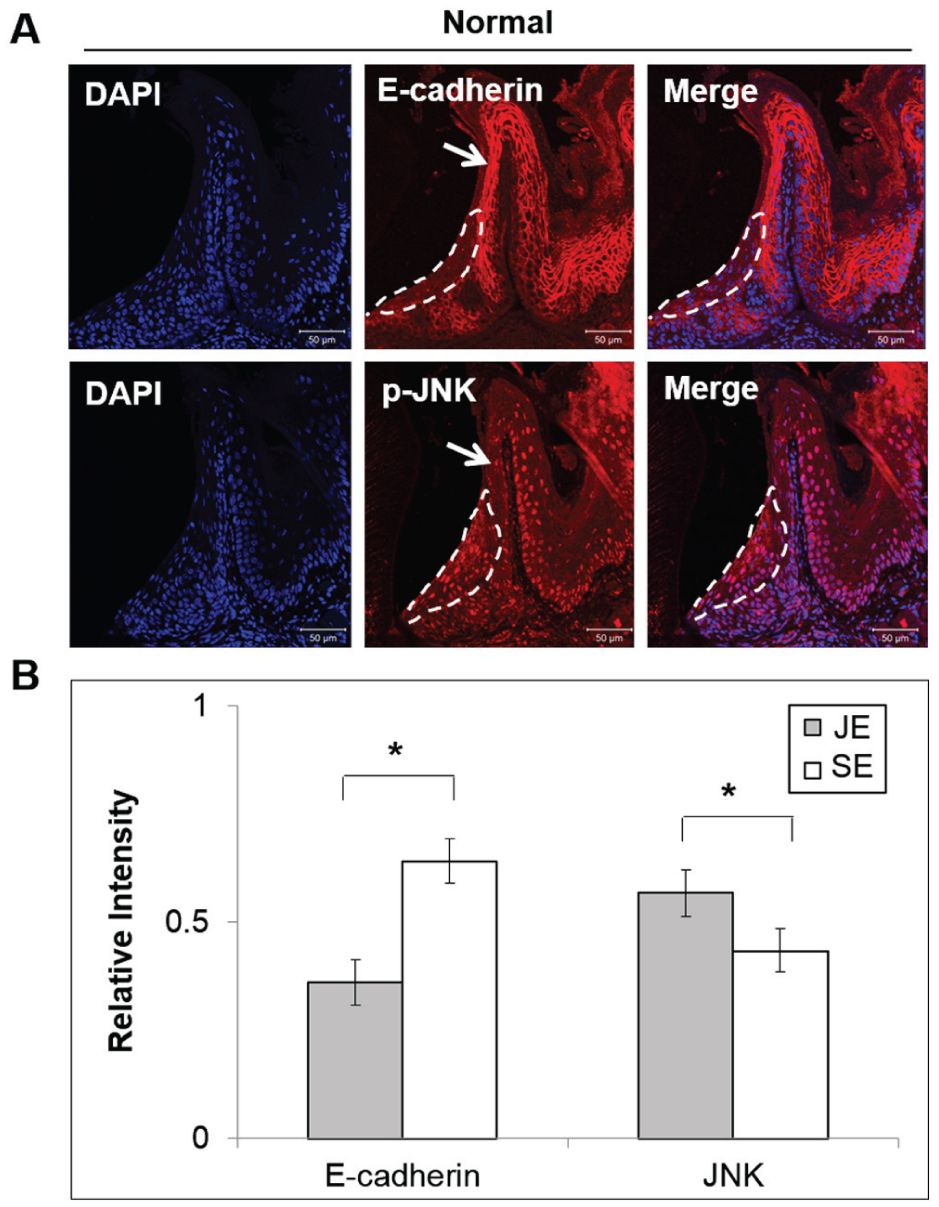
p-JNK is highly expressed within the intercellular junctions of the mouse junctional epithelium, whereas E-cadherin is poorly expressed. (
In Vitro Formation of the Intercellular E-Cadherin Junctions between Human Gingival Epithelial Cells Depends on the Stiffness of the Underlying Surface
The JE is unusual in that it attaches to the surface of enamel, which is the hardest surface in the human body. Attachment to this surface may be enabled through special characteristics of the JE not found in other epithelia. Intercellular junctions between human gingival epithelial cells were compared after culturing on flexible collagen gel or in solid polystyrene culture dishes, both of which were coated with a high concentration of fibronectin, which disrupts the intercellular junctions. Interestingly, cells cultured on the collagen gels maintained their E-cadherin junctions (Fig. 5A). However, HOK-16B cells cultured on the stiff surface of the culture dishes lost their intercellular connections. These results indicate that a stiff surface may induce the activation of a RhoA-JNK signaling pathway that leads to the disruption of the intercellular E-cadherin junctions. As expected, the level of active phosphorylated JNK was higher in cells grown on the stiff culture dishes compared with those grown on the softer collagen surface (Fig. 5B). JNK activation by anisomycin disrupted the intercellular E-cadherin junctions formed between cells on the collagen gel surfaces. In contrast, JNK inhibition by SP600125 restored these junctions on the stiff culture dishes. Because RhoA activity could not be directly analyzed due to the cytoskeletal modifications that would occur during cell detachment from the gel matrix, lower RhoA activity on the soft gel surfaces was confirmed by examining the phenotypical changes induced when activating RhoA. RhoA activation by CNF-1 disrupted the intercellular E-cadherin junctions between cells grown on the collagen gel surface. In contrast, RhoA inhibition by C3 restored the junctions between cells grown in the stiff culture dishes. These results suggest that the stiffness of the surface to which epithelial cells attach and grow may reduce the stability of the intercellular junctions between cells through activation of RhoA-JNK signaling. Thus, it is highly probable that intercellular junctions within the JE are poorly formed as a result of the underlying, highly solid enamel surface.
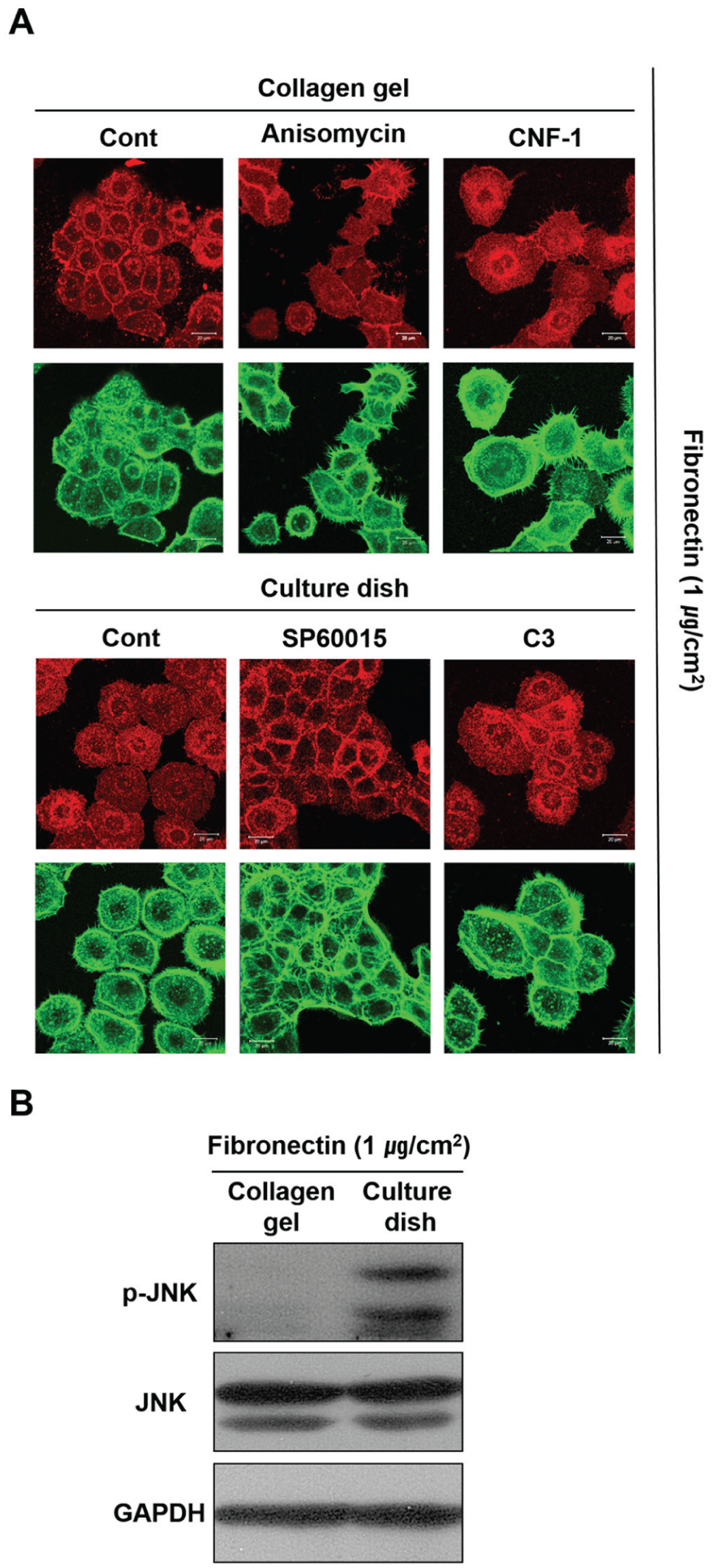
Intercellular E-cadherin junctions between HOK-16B cells are poorly formed due to the upregulation of p-JNK that results from growth on solid surfaces. (
Discussion
The present study demonstrates that the formation of cell-to-cell junctions between human gingival epithelial cells is regulated by cell signaling pathways. Therefore, the structural characteristics of the JE may be derived from differences in the signaling pathways controlling the intercellular junctions. The JE of mouse molars contained high JNK activity accompanied by poorly formed E-cadherin junctions. This inverse relationship between the development of the intercellular junctions and JNK activity was apparent in vitro. JNK activation downregulated E-cadherin expression at the site of the disrupted cellular junctions. These results strongly suggest that the limited development of intercellular E-cadherin junctions within the JE may be a result of high JNK activity, which may result in wide intercellular gaps that allow the migration of protective leukocytes into the epithelium. However, the destruction of cell-to-cell junctions in the gingival epithelial cells may lead to the deterioration of the barrier provided by the JE, which protects it from bacterial invasion or the diffusion of toxic substances. It would ultimately lead to infection or inflammation of the connective tissue underlying the epithelium. It has been reported that E-cadherin expression decreases further within the pocket epithelium compared with the healthy JE (Ye et al. 2000). These results indicate that JNK may be a master signaling molecule regulating the intercellular junctions within the JE. Furthermore, it is possible that JNK inhibition may modulate gingivitis and periodontitis by promoting the strengthening of these intercellular junctions.
The JE is peculiar in that it has 2 basal laminae, one opposing the gingival connective tissue and the other opposing the solid enamel surface. Interestingly, the JE interfaces with enamel, which is the stiffest surface in the human body. Thus, we hypothesized that the solid enamel surface may promote dissociation of E-cadherin junctions within the JE. To this end, we compared the cellular junctions between human gingival epithelial cells cultured on stiff culture dishes to those formed on soft collagen gels. Dissociation of human gingival epithelial cells from the culture dishes strongly points to the poor formation of intercellular E-cadherin junctions. Furthermore, this dissociation was associated with higher JNK and RhoA activities compared with the confluent layer of cells formed on the soft collagen gels. Based on these results, we conclude that the stiffness of a surface leads to the upregulation of RhoA, which in turn activates JNK, resulting in a low number of intercellular junctions within the JE. The poor formation of adherens junctions between human skin keratinocytes grown on stiff surfaces further supports our hypothesis (You et al. 2013). Studies using a conditional JNK or RhoA knockdown animal model will be required to confirm this hypothesis.
The finding that RhoA plays a role in regulating the intercellular junctions between gingival epithelial cells by activating JNK is notable because various bacterial toxins regulate RhoA activity. Therefore, it is likely that the E-cadherin junctions within the JE may be further destroyed if bacterial toxins activate RhoA-JNK signaling. Although it is unknown whether oral bacteria regulate RhoA, it is well known that many bacteria secrete toxins that affect RhoA activity. CNF-1, used to activate RhoA in this study, is a 115-kDa toxin that is produced by uropathogenic Escherichia coli (Rippere-Lampe et al. 2001). However, the regulation of E-cadherin junctions by bacterial toxins may be complicated by the bacterial toxins downregulating RhoA activity, which may prevent the disruption of E-cadherin junction. Exoenzyme C3, used in the present study to downregulate RhoA activity, is a 26-kDa molecule with ADP-ribosyltransferase activity that is produced by various bacteria, including Staphylococcus aureus (Hauser et al. 1993).
Tight junction (TJ) may contribute to the structure of the intercellular junction of the JE in addition to the E-cadherin. However, there was no typical TJ developed in the HOK-16B cells. ZO-1, an adaptor molecule of the TJ, was scattered throughout the whole cytoplasm rather than being confined to develop the TJ at the cell membrane (Appendix Fig. 5 and Choi et al. 2013). This result confirms that the TJ, if any, contributes much less to the formation of the physical intercellular junction than the E-cadherin junctions do in the gingival epithelium (Damek-Poprawa et al. 2013).
In summary, the results of the present study suggest that RhoA-JNK signaling may regulate the structural characteristics of the intercellular junctions of human gingival epithelial cells. Moreover, inhibiting the RhoA-JNK signaling axis may ultimately strengthen those intercellular junctions. Further studies of this signaling axis may explain why the JE contains cells with wide intercellular junctions and scarce E-cadherin content and may suggest strategies for regulating these junctions to protect the tooth-supporting gingiva from the bacteria in the oral cavity.
Author Contributions
G. Lee, contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; H.J. Kim, contributed to design, data acquisition, and analysis, drafted and critically revised the manuscript; H.-M. Kim, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry for Health & Welfare, Republic of Korea; contract grant number: HI13C1709.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.