
Abstract
Craniofacial development in vertebrates involves the coordinated growth, migration, and fusion of several facial prominences during embryogenesis, processes governed by strict genetic and molecular controls. A failure in any of the precise spatiotemporal sequences of events leading to prominence fusion often leads to anomalous facial, skull, and jaw formation—conditions termed craniofacial defects (CFDs). Affecting approximately 0.1% to 0.3% of live births, CFDs are a highly heterogeneous class of developmental anomalies, which are often underpinned by genetic mutations. Therefore, identifying novel disease-causing mutations in genes that regulate craniofacial development is a critical prerequisite to develop new preventive or therapeutic measures. The Grainyhead-like (GRHL) transcription factors are one such gene family, performing evolutionarily conserved roles in craniofacial patterning. The antecedent member of this family, Drosophila grainyhead (grh), is required for head skeleton development in fruit flies, loss or mutation of Grhl family members in mouse and zebrafish models leads to defects of both maxilla and mandible, and recently, mutations in human GRHL3 have been shown to cause or contribute to both syndromic (Van Der Woude syndrome) and nonsyndromic palatal clefts. In this review, we summarize the current knowledge regarding the craniofacial-specific function of the Grainyhead-like family in multiple model species, identify some of the major target genes regulated by the Grhl transcription factors in craniofacial patterning, and, by examining animal models, draw inferences as to how these data will inform the likely roles of GRHL factors in human CFDs comprising palatal clefting. By understanding the molecular networks regulated by Grhl2 and Grhl3 target genes in other systems, we can propose likely pathways that mediate the effects of these transcription factors in human palatogenesis.
Keywords
Introduction
Recent work shows that the Grainyhead-like (Grhl) transcription factors are critical, highly conserved regulators of craniofacial development, regulating formation of both upper and lower jaws (maxilla and mandible, respectively). Importantly, these factors demonstrate clear clinical relevance, underpinning several human disorders of the craniofacial complex, particularly palatal clefting (both syndromic and nonsyndromic), otic development, age-related hearing defects, and cognitive disorders. In this review, we discuss the role of the Grhl family in vertebrate craniofacial development, review the nature and identity of genetic networks involving the Grhl factors, and provide a summary of the numerous genetic models that have been generated to explore the role of Grhl genes in embryogenesis.
A Brief Overview of Craniofacial Development
Vertebrate facial formation predominantly comprises the development, migration, outgrowth, and fusion of 5 prominences: the frontonasal prominence (FNP), which forms the facial midline and nose, and the paired maxillary (MXP) and mandibular (MDP) prominences of the first pharyngeal arch (PA1). In mouse, the PA1 arises at embryonic day (E) 8.25 (~week 4 in humans; Yoon et al. 2000) and segregates into the MXP and MDP. The MDP consists of paired processes that grow toward the midline and merge; MDP development is subsequently driven by mesenchymal proliferation and anterior migration and elongation of the developing mandible (Abramyan and Richman 2015). As development proceeds, the FNP is divided into the medial and lateral nasal prominences (MNP, LNP) by the nasal pits (Bush and Jiang 2012; Dworkin et al. 2016). At E10.5 (35 d in human), the MNP and LNP have started to fuse rostral to the nasal pits; fusion commences at the posterior nasal pit and proceeds in an anterior direction. At E11.0 (38 d in human), the MXP and MNP have started to fuse. The epithelial seam between the fused primordia undergoes apoptosis to allow mesenchymal confluence and complete upper lip formation by E12.5 (48 d in human). The intermaxillary segment then outgrows into the oral cavity to form the anterior or “primary” part of the palate.
Once upper lip morphogenesis is complete, the alae of the nose are derived from the LNP while the upper lip is derived from the MNP and MXP. Although the LNP does not contribute to the lip, a failure of fusion between the LNP and MNP causes a cleft in the lip that extends into the nostril. As failure of lip fusion often disrupts fusion between the secondary palatal shelves, which occurs much later in embryonic development, cleft lip is often accompanied by cleft palate. The palatal shelves start to grow (first vertically, then horizontally) from the MXP at E11.5, elevate, and eventually merge at E15.0, giving rise to the secondary palate. Fusion completes by E17.0, after the seam of medial edge epithelium (MEE) between the shelves dissolves by migration, apoptosis, and epithelial-mesenchymal transition (EMT). The secondary palate also fuses with the primary palate and the nasal septum to form the roof of the mouth (~weeks 8 to 12 in humans; Diewert 1983; Yoon et al. 2000). Aberrant fusion leads to disorders, including cleft lip and palate (CL/P), cleft lip only (CL), or cleft palate only (CPO) (Wilkie and Morriss-Kay 2001).
Both ectoderm- and endoderm-derived epithelia, as well as neural crest cells (NCCs), contribute to craniofacial patterning and morphogenesis (Dworkin et al. 2016). In addition, a transient, supraepithelial structure, the periderm, is critical in the prevention of oral clefts. This single-cell thickness layer arises from proliferating epithelia in the midgestation embryo, such as intestine, limb buds, and palatal shelves (M’Boneko and Merker 1988), provides a protective “coating” for these tissues, and persists until shortly before birth. Oral periderm prevents premature adhesion between closely appositioned epithelia within the oral cavity, such as the palatal shelves, mandible, and tongue (Lan et al. 2015). Importantly, defects in oral periderm development and/or maintenance underpin palatal clefting in numerous mouse models (Ingraham et al. 2006; Richardson et al. 2009; Peyrard-Janvid et al. 2014; Richardson et al. 2014).
Understanding the genetic and molecular regulation of these myriad developmental processes will identify both risk factors and drive discovery of potential therapeutic targets.
Insights into the grh/grhl Family from Nonmammalian Species—Drosophila and Zebrafish
The Grainyhead family diverged from related transcription factors when multicellular animals first evolved epithelia (Traylor-Knowles et al. 2010). The antecedent member of this family, grainyhead (grh), was discovered in fruit fly (Drosophila) as a key regulator of epidermal integrity, tissue specification, pro/antiproliferative regulation of neuroblast fate, dorsal hole closure, and epidermal denticle polarity (Bray and Kafatos 1991; Cenci and Gould 2005; Maurange et al. 2008). Both grh and vertebrate orthologues, Grainyhead-like (Grhl) proteins, comprise an amino-terminal transcriptional activation domain (TAD), central DNA-binding domain, and carboxyl-terminal dimerization domain (Wilanowski et al. 2002). Grh/Grhl factors bind to the conserved DNA sequence AACCGGTT within noncoding regulatory regions of target genes (Ting, Caddy, Hislop, et al. 2005; Boglev et al. 2011), with subsequent activating/repressive function dictated by the nature of subsequent homo- and/or heterodimerization (Attardi et al. 1993; Wilanowski et al. 2002; Ting, Wilanowski, Cerruti, et al. 2003; Nevil et al. 2017).
grainyhead was named for the grainy and discontinuous morphology of the chitinous “head-skeleton” in Drosophila larvae with grh mutations (Bray and Kafatos 1991). In grh-null flies, both dorsal and ventral arms of the craniofacial apparatus are smaller, globular, and distended. The anterior-most mouth hooks are also significantly smaller. Although an invertebrate, the head-skeleton tissue in flies shares both structural and functional homology with vertebrates, and many genes that regulate Drosophila head-skeleton patterning also regulate vertebrate craniofacial development, such as msh/Msx (Alappat et al. 2003), odd-skipped/Osr2 (Lan et al. 2001), and otd/Otx2 (Montalta-He et al. 2002), confirming the relevance of this model.
In vertebrates, the role of grh is subfunctionalized into 3 orthologues—Grhl1, Grhl2, and Grhl3. Inactivation of vertebrate Grhl genes leads to developmental defects, including the craniofacial skeleton (Ting, Wilanowski, Auden, et al. 2003; Ting, Caddy, Hislop, et al. 2005; Yu et al. 2009; Rifat et al. 2010; Werth et al. 2010; Boglev et al. 2011; Pyrgaki et al. 2011; Dworkin et al. 2014; Menke et al. 2015). Numerous insights into the roles of vertebrate grhl genes have been gained from zebrafish (Danio rerio), owing predominantly to its high genetic tractability, rapid development, and optical clarity of embryos. Unlike mice and humans, zebrafish comprise 4 grh orthologues—grhl1, grhl2a, grhl2b, and grhl3 (Janicke et al. 2010; Dworkin et al. 2012)—and numerous morpholino (MO)–mediated grhl knockdown studies have been reported. Despite robust expression throughout the developing skin and pharyngeal arches, grhl1 does not appear to play a significant role in craniofacial development (Janicke et al. 2010); however, MO-mediated grhl1 knockdown led to developmental malformations of the inner ear (Liu et al. 2015). Similarly, grhl2a and grhl2b loss-of-function models do not suggest involvement in craniofacial development. grhl2a is maternally deposited and ubiquitously expressed (Janicke et al. 2010; Dworkin et al. 2012), yet MO-mediated knockdown of grhl2a did not yield discernible phenotypes (Dworkin et al. 2012). grhl2b is expressed in the polster, anterior neural plate, trunk neural crest, and otic precursors from approximately 8 h postfertilization (hpf) (Dworkin et al. 2012). Both genomic deletion (Han et al. 2011) and MO-mediated knockdown (Dworkin et al. 2012) of grhl2b led to defects in otic and midbrain-hindbrain boundary formation, respectively, although the facial skeleton was not affected. Concurrent MO-mediated knockdown of both grhl2a and grhl2b led to defects in axial formation, notochord formation, and convergence-extension mediated migration up to 24 hpf (Dworkin et al. 2012), although the effect that cooperative abrogation of grhl2a/b function may have on subsequent development of the craniofacial skeleton has not been explored.
Conversely, however, the role of grhl3 in the development, patterning, and growth of the fish craniofacial structures has been described (Dworkin et al. 2014). grhl3 is initially expressed in the enveloping layer (EVL) during the onset of zygotic transcription (~4.5 hpf). It is then rapidly downregulated as epiboly and gastrulation proceed, before strong and specific reexpression within the medial endodermal epithelium of the pharynx from ~18 hpf. grhl3 remains expressed within the epithelium of the pharyngeal arches until at least 96 hpf (Dworkin et al. 2014). MO-mediated knockdown of grhl3 led to significant hypoplasia of the lower jaw cartilage, with concomitant defects in the articulation of the jaw joint with the palatoquadrate, reduction of both the basihyal and ceratohyal bones, and frequent absence or hypomorphism of the cerato-branchials, although maxillary structures were not compromised. Our analyses identified that the soluble signaling factor endothelin-1 (edn1), itself expressed in the oral epithelium and implicated in morphogenesis and extension of the lower jaw (Kurihara et al. 1997; Miller et al. 2000; Miller and Kimmel 2001), was a direct target of grhl3 within the pharyngeal endoderm. As grhl3 is not expressed within the NCCs, which migrated and homed to the arches correctly (Dworkin et al. 2014), these data indicate a disruption of correct signaling from the epithelium to the NCC-derived mesenchyme, resulting in secondary (non–cell autonomous) NCC death and deficient mandibular outgrowth (Dworkin et al. 2014).
Grhl Mutant Mouse Models
The expression of murine Grhl genes closely approximates that of zebrafish. All 3 orthologues are extensively expressed within the oral epithelium (Auden et al. 2006); Grhl2 is the earliest orthologue expressed in FNP, MXP, and MDP epithelium from E10.5 (Fig. 1). As in zebrafish, Grhl1 appears dispensable for craniofacial patterning (Wilanowski et al. 2008). However, numerous mutant mouse lines reveal distinct roles for Grhl2 and Grhl3 in craniofacial development (Table). The role of Grhl2 in craniofacial development has been difficult to determine from mouse models, due to early embryonic lethality. To date, 5 separate murine deletion models of Grhl2 have been generated, all of which present with cleft face, neural tube defects, and embryonic lethality by E9.5 to E11.5 (Rifat et al. 2010; Werth et al. 2010; Brouns et al. 2011; Pyrgaki et al. 2011; Menke et al. 2015). Scanning electron microscopy (SEM) data show that Grhl2tm1.1Jane null embryos present with hypomorphic maxillary prominences by E11.5, approaching the time of lethality, in addition to an open cranial and caudal neural tube (Fig. 1). Consistent with smaller MXPs, embryos from 2 lines, however, occasionally survive to E18.5 (Grhl2m1Nisw/m1Nisw and Grhl2clf3/clf3), presenting with hypomorphic and split maxillae, as well as short snouts. Midline craniofacial malformations have also been associated with reduced Grhl2 expression in Tuft mice (Fong et al. 2016), owing to a mutation in methylcytosine dioxygenase, suggesting Grhl2 may also underpin these malformations. More generally, murine data suggest that Grhl2 may be a conserved regulator of midline convergence and/or fusion. Whether the mechanisms involved are specific to the palatal shelves or are a secondary consequence of generalized defects in the establishment of axial symmetry remains to be determined.
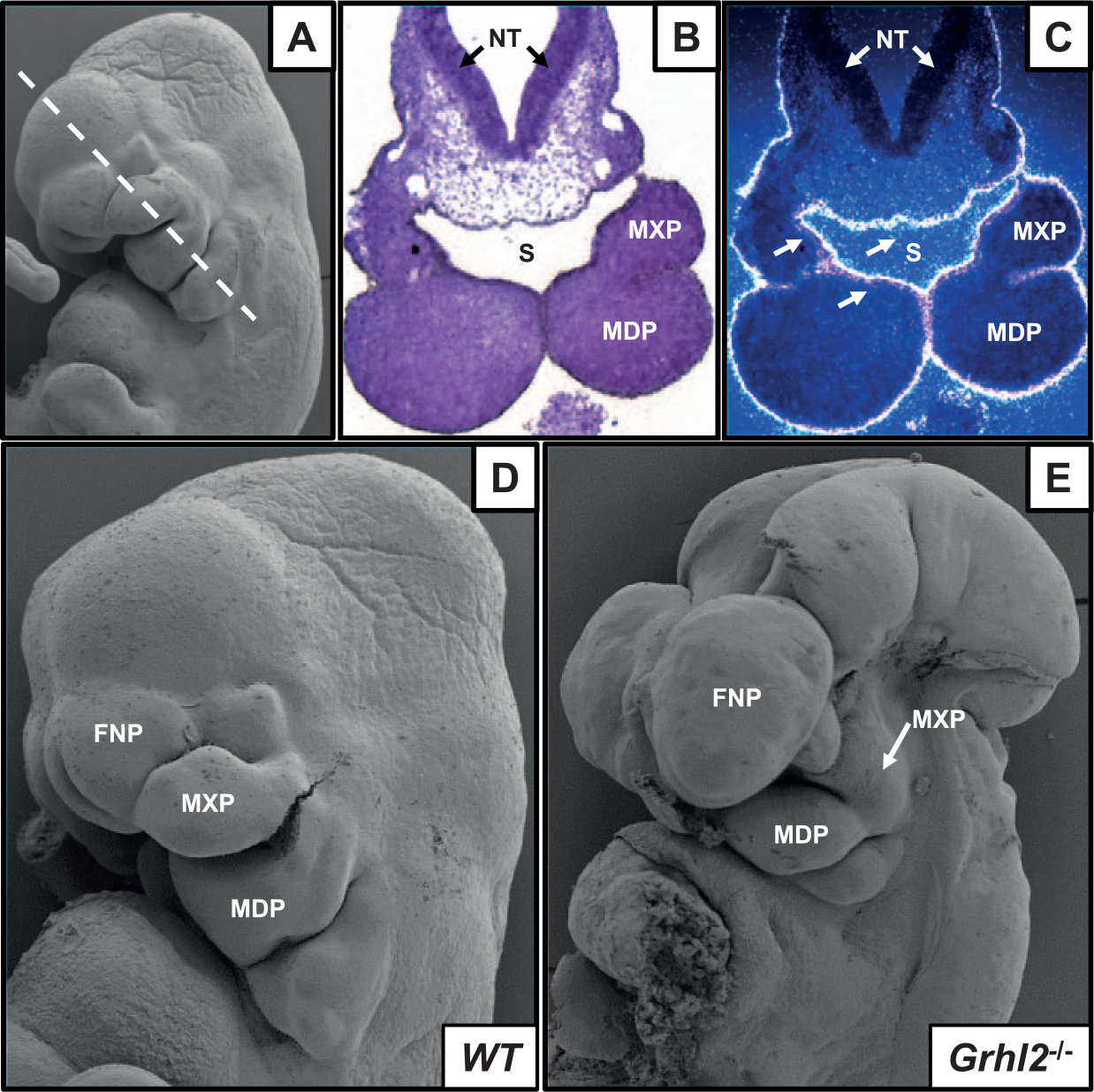
Grhl2 expression and function in craniofacial primordia. (
Murine Models of Grhl2 and Grhl3 Loss and Misexpression.
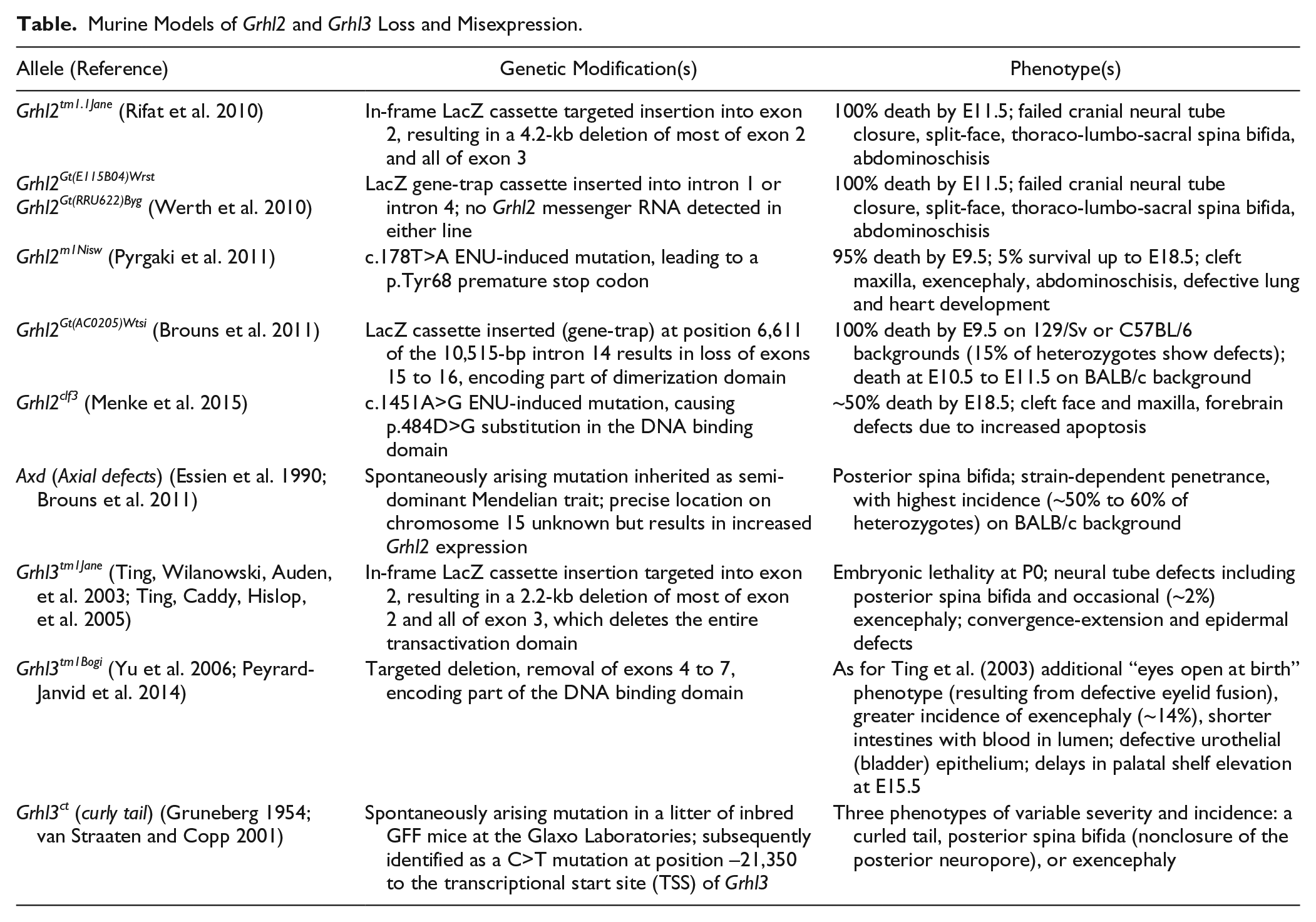
Two Grhl3 “null” mouse mutations have been generated using gene-targeting approaches: Grhl3tm1Jane, which removes the entire TAD from Grhl3 (Ting, Wilanowski, Auden, et al. 2003), and Grhl3tm1Bogi, which removes parts of the TAD and DNA-binding domain (DBD) (Yu et al. 2006). Grhl3tm1Jane/tm1Jane embryos died soon after birth from dehydration (Ting, Caddy, Hislop, et al. 2005). They also presented with a short and squat body shape, spina bifida, and prematurely apposed skull bones (Ting, Wilanowski, Auden, et al. 2003; Goldie et al. 2016). These embryos did not display cleft palate at E18.5 but did display delays in palatal shelf elevation at earlier stages of development (our unpublished observations). Similarly, Grhl3tm1Bogi/tm1Bogi embryos presented with bilateral adhesions of the palate to the tongue and partially penetrant (17%) delays in palatal shelf elevation (analogous to an embryonic “cleft”) at E15.5 (Peyrard-Janvid et al. 2014). These embryos presented with defects in the oral periderm, which overlies the palatal epithelium, resulting in pathogenic adhesion of the palatal shelves to the developing mandible and/or tongue (Peyrard-Janvid et al. 2014). It is possible that the Grhl3tm1Bogi allele inhibits Grhl2 in a dominant negative manner to induce the more severe craniofacial phenotype in this line. This is supported by other aspects of the phenotype being more severe in Grhl3tm1Bogi/tm1Bogi embryos than in Grhl3tm1Jane/ tm1Jane embryos, including a higher incidence of exencephaly, eyes open at birth (due to defective eyelid development), and shorter, blood-filled intestines.
Grhl2 and Grhl3 may have partially co-redundant roles in craniofacial development, suggested by overlapping roles in neural tube closure. Specifically, Grhl2 or Grhl3 can both mediate closure of parts of the neural tube, with gene dosage being the key factor that determines whether closure is successful (Rifat et al. 2010). Other evidence suggests that Grhl2 and Grhl3 have overlapping target gene specificities. Expression of Grhl2 as a knock-in allele from the Grhl3 locus partially rescued neural tube defects associated with Grhl3 nullizygosity (Rifat et al. 2010). The potential for heterodimer formation complicates interpretation of phenotypes arising from overexpression/mutation of Grhl genes. For example, increased expression of Grhl2 in the Axd spontaneous mutant mouse is associated with neural tube defects similar to those of Grhl3tm1Jane/tm1Jane mice, although the Axd mice themselves do not present with craniofacial defects. However, the Axd phenotype is rescued by normalizing Grhl2 levels (Brouns et al. 2011). This may indicate that excess Grhl2 in the Axd mouse heterodimerizes with Grhl3 and disrupts its activity. Alternatively, a Grhl2/Grhl3 heterodimer may be required for neural tube closure; increased Grhl2 expression in the Axd mouse may disrupt its formation.
These observations indicate that whereas Grhl3 is required in the palatal epithelium/periderm to prevent premature adhesion (possibly in conjunction with Grhl2), Grhl2 plays an earlier, broader role in maxillary process outgrowth and/or palatal shelf elevation. Thus, palatal clefting associated with Grhl2 and Grhl3 mutations is likely to be a consequence of maxillary/midline growth deficiency and disruption of epithelial integrity, respectively (Fig. 2), indicative of complementary roles in formation of the craniofacial complex.
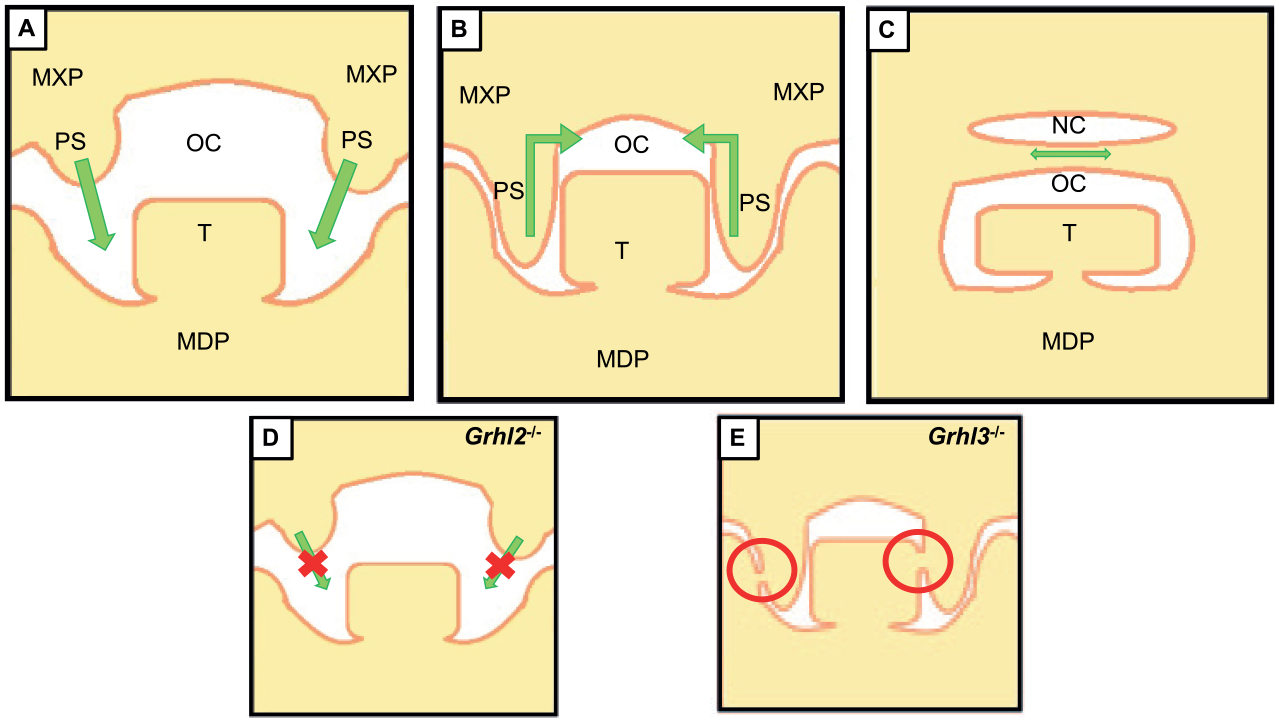
Roles of Grhl2 and Grhl3 in palatogenesis. (
GRHL Genes in Human Craniofacial Disorders
GRHL2 is a clinically relevant gene in defects pertaining to the head and face. Recessive missense mutations in GRHL2 cause ectodermal dysplasia (Petrof et al. 2014), a disorder characterized by pigmentation of oral mucosa and/or tongue and abnormal dentition, including hypodontia and enamel hypoplasia. More deleterious frameshift and missense mutations in GRHL2 are found in the heterozygous but not homozygous state and cause dominant, progressive hearing loss (Peters et al. 2002; Vona et al. 2013). Furthermore, a single-nucleotide polymorphism (SNP) in intron 1 of GRHL2 increases the risk of developing age-related hearing loss at the population level (Van Laer et al. 2008). Grhl2 is expressed in the epithelial cells lining the central duct of the inner ear or cochlea (Peters et al. 2002); maintenance of this epithelium may be compromised by GRHL2 haploinsufficiency. Last, 6 patients with facial dysmorphisms and cognitive deficits, harboring microdeletions at chromosomal region 8q22.2q22.3, have been identified. These patients are hemizygous for a 1.9-Mb region containing 9 genes, of which only GRHL2 has been implicated in craniofacial abnormalities. It is tempting to speculate that hemizygosity for GRHL2 may be the underlying genetic disturbance underpinning these defects (Kuechler et al. 2011; Sinajon et al. 2015). Although not yet implicated in palatal clefting in humans, this is likely due to complete (nullizygous) loss-of-function mutations in humans being incompatible with life.
Recent work has identified GRHL3 mutations in the etiology of human spina bifida (Lemay et al. 2017) and, importantly, also syndromic (Van der Woude syndrome [VWS]; Peyrard-Janvid et al. 2014) and nonsyndromic (Leslie et al. 2016; Mangold et al. 2016) palatal clefts. In the majority of cases (~70%), VWS is caused by mutations in the IRF6 gene (Kondo et al. 2002), the most common syndromic form of orofacial clefting (Rizos and Spyropoulos 2004); importantly, IRF6 is thought to be a regulator of GRHL3 (discussed in the next section). However, mutations in GRHL3 have been implicated in ~5% of VWS cases with no mutations in IRF6 (Koillinen et al. 2001), suggesting that these 2 genes operate within a common, linear pathway. GRHL3 mutations also segregate with nonsyndromic CPO (Mangold et al. 2016). GRHL3 mutations with predicted severe consequences on protein function are not found in the homozygous or compound heterozygous state and are likely not compatible with life. A common missense variant in GRHL3 also increases the risk of CPO at the population level (Leslie et al. 2016; Mangold et al. 2016). This dominant allele encodes a p.Thr454Met amino acid substitution within the DNA binding domain and reduces transactivation activity to 40% of wild-type in oral epithelial cells. Furthermore, the p.Thr454Met variant inhibits transactivation by coexpressed wild-type GRHL3, suggesting that it has dominant negative activity.
IRF6-GRHL3 Transcriptional Network and Clefting—A Common Pathway?
The IRF6 locus is genetically linked to the risk of orofacial clefting (Zucchero et al. 2004). Numerous lines of evidence indicate that the IRF6-GRHL3 network comprises a critical regulatory pathway in the etiology of palatal clefts. IRF6 binds to the GRHL3 promoter in human keratinocytes, and RNA interference (RNAi)–mediated knockdown of IRF6 leads to downregulation of GRHL3 in these cells (Botti et al. 2011; Kwa et al. 2014). Both Irf6 and Grhl3 are expressed within the palatal epithelium (Auden et al. 2006; Peyrard-Janvid et al. 2014), as well as the pharyngeal arches of zebrafish (Ben et al. 2005). Irf6 mutant mouse embryos are unable to complete epidermal differentiation, leading to skin thickening at e17.5, abnormal persistence of desmosomes, and failure of the skin barrier to form (Ingraham et al. 2006; Richardson et al. 2006), similar defects to those seen following inactivation of Grhl3 (Ting, Caddy, Hislop, et al. 2005; Ting, Caddy, Wilanowski, et al. 2005). Like Irf6–/– embryos, Grhl3tm1Bogi/tm1Bogi embryos showed abnormal periderm differentiation. Furthermore, the medial edge epithelium dissolved during palatal shelf fusion in Grhl3tm1Bogi/tm1Bogi embryos but not in Irf6R84C/R84C or Irf6–/– embryos (Richardson et al. 2009; Peyrard-Janvid et al. 2014). Oral adhesions and periderm defects are more severe in Irf6–/– embryos than in Grhl3–/– embryos (Peyrard-Janvid et al. 2014), possibly due to the loss of Grhl3 expression in oral periderm in Irf6–/– mouse embryos at e14.5, exacerbating the Irf6–/– phenotype (de la Garza et al. 2013). These data suggest that Irf6 is absolutely required for periderm differentiation, while a hypomorphic periderm may form in the absence of Grhl3. Interestingly, Grhl3+/–Irf6+/– mouse embryos do not have palate defects (Peyrard-Janvid et al. 2014), suggesting either that double heterozygosity is insufficient to result in a phenotype or possibly that other genes within the gene-regulatory network (GRN) may compensate for this loss. However, given the high degree of similarity between mouse and human palatogenesis (Lan et al. 2015), a loss of periderm is the likely pathological basis for facial clefting in humans carrying GRHL3 mutations.
The zebrafish has proven useful in characterizing molecular consequences of human GRHL3 mutations. An ideal fish model would harbor the identical genetic lesions identified in human GRHL3 within the fish grhl3 locus (a possibility due to the advent of CRISPR/Cas9 technology) to specifically identify consequences on craniofacial development. One caveat is that grhl3 in fish palatogenesis has not yet been described, although strong conserved parallels exist in palatogenic gene programs between fish and mammals (Swartz et al. 2011). However, a surrogate model has been developed, using the striking cellular molecular, functional, and genetic similarities between the zebrafish EVL and the mammalian oral periderm (de la Garza et al. 2012). Both of these are transient, simple, squamous epithelium–derived layers (Fig. 3) that act as a permeability barrier with the surrounding environment. Notably, keratins and Irf6 and Grhl genes are expressed in oral periderm and zebrafish EVL (de la Garza et al. 2013; Janicke et al. 2010; Dworkin et al. 2014), suggesting that similar genetic mechanisms may operate within both structures. Zebrafish embryos injected with RNA encoding a dominant negative form of Irf6 exhibit reduced grhl3 expression in EVL (de la Garza et al. 2013), and an evolutionarily conserved element in mouse Grhl3 intron 15 has enhancer activity in zebrafish EVL that is dependent on an Irf6 binding site.
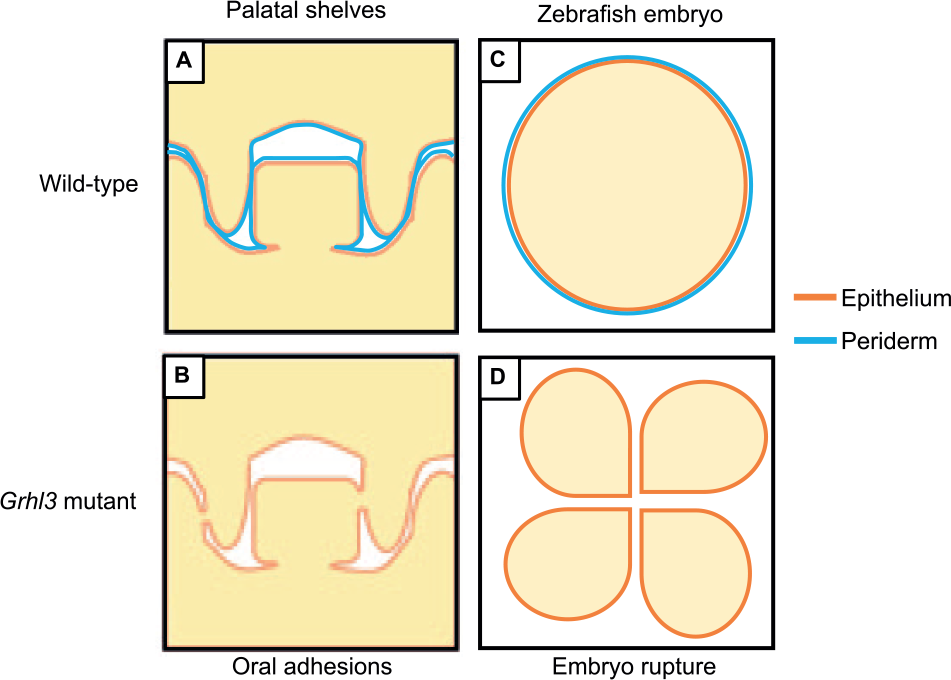
Role of oral periderm in palate and zebrafish development. (
The zebrafish has also been successfully used to determine whether GRHL3 mutations identified in VWS were nullizygous or resulted in dominant-negative GRHL3 function. Transient overexpression assays exhibit peak expression at epiboly; analysis of early embryonic events that are disrupted is indicative of gene function. When transiently overexpressed in zebrafish embryos, dominant-negative grhl3 led to EVL rupture during epiboly (de la Garza et al. 2012). Injection of mutant but not wild-type GRHL3 constructs comprising 4 missense or 1 frameshift mutation resulted in stalling of epiboly and embryonic rupture of the EVL at approximately 8 hpf, suggesting a dominant-negative action (Peyrard-Janvid et al. 2014). Importantly, the embryo-rupture effect was cell autonomous. Coinjecting GRHL3 and biotin into individual cells of the 16-cell stage embryo showed that dominant-negative GRHL3 led to downregulation of the EVL marker krt4 only within individual injected cells (and not adjacent, noninjected cells; Peyrard-Janvid et al. 2014). This assay was also used to determine whether the p.Thr454Met GRHL3 variant that mediates susceptibility to CPO has a dominant negative effect (Leslie et al. 2016). Injection of complementary DNA (cDNA) encoding this variant also caused embryo rupture and reduced expression of krt4, indicating dominant-negative activity in this system. These data suggest that grhl3 in fish is required for the development, maintenance, and structural integrity of simple epithelia, and this model may provide significant information as to the GRN regulated by irf6/grhl3 within other such epithelia, such as the oral periderm of mammals. It also supports the idea that GRHL3 is part of a critical signaling pathway in the etiology and pathogenesis of palatal clefts caused by a disruption or disintegration of the oral periderm.
Further Transcriptional Targets of grh/grhl/Grhl Genes in Craniofacial Development?
Other molecular pathways by which Grhl2/Grhl3 exert their effects on palate development are not fully understood. Few upstream regulatory factors of the grh/Grhl family have been identified, although both branchless/FGF8 (Hemphala et al. 2003; Dworkin et al. 2012) and TET1 (Fong et al. 2016) influence grh/Grhl expression and are important for craniofacial development (Trumpp et al. 1999). However, the downstream targets of this family are beginning to be well characterized through multiple large-scale screening approaches (Aue et al. 2015; Gao et al. 2015; Pifer et al. 2016; Nevil et al. 2017), as well as targeted analysis of candidate genes. Grhl2 maintains epithelial cell identity of kidney, breast, and potentially surface ectoderm by directly transactivating Cldn4, Cdh1, Ovol2 (Werth et al. 2010; Aue et al. 2015; Chung et al. 2016), and Mir200, a microRNA that represses Zeb1 and Zeb2 (Gregory et al. 2008). Cldn4, a member of the claudin family of tight junction proteins, is expressed within the oral periderm during development and within the MEE at the point of midline adhesion, suggesting it is important for maintenance of structural integrity during fusion (Dravis and Henkemeyer 2011; Yoshida et al. 2012). Cdh1 (E-cadherin) is a well-validated Grhl2 target (Werth et al. 2010; Pyrgaki et al. 2011; Quan et al. 2015), which has also been implicated in human palatal clefting, possibly due to MEE breakdown and/or aberrant EMT (Frebourg et al. 2006; Nawshad et al. 2007). Grhl2 is a potent EMT suppressor via numerous pathways, such as Ovol1/2 (Aue et al. 2015; Yoh and Prywes 2015), Zeb1/2 (Cieply et al. 2012; Mooney et al. 2017), TGFβ, and MMP2 (Xiang et al. 2017). It is possible that a similar regulatory axis may exist within the palate, allowing Grhl2 to maintain MXP epithelial cellular identity and consequently provide regulatory signals to the MXP mesenchyme, which influence subsequent shelf outgrowth and elevation (Fig. 4).
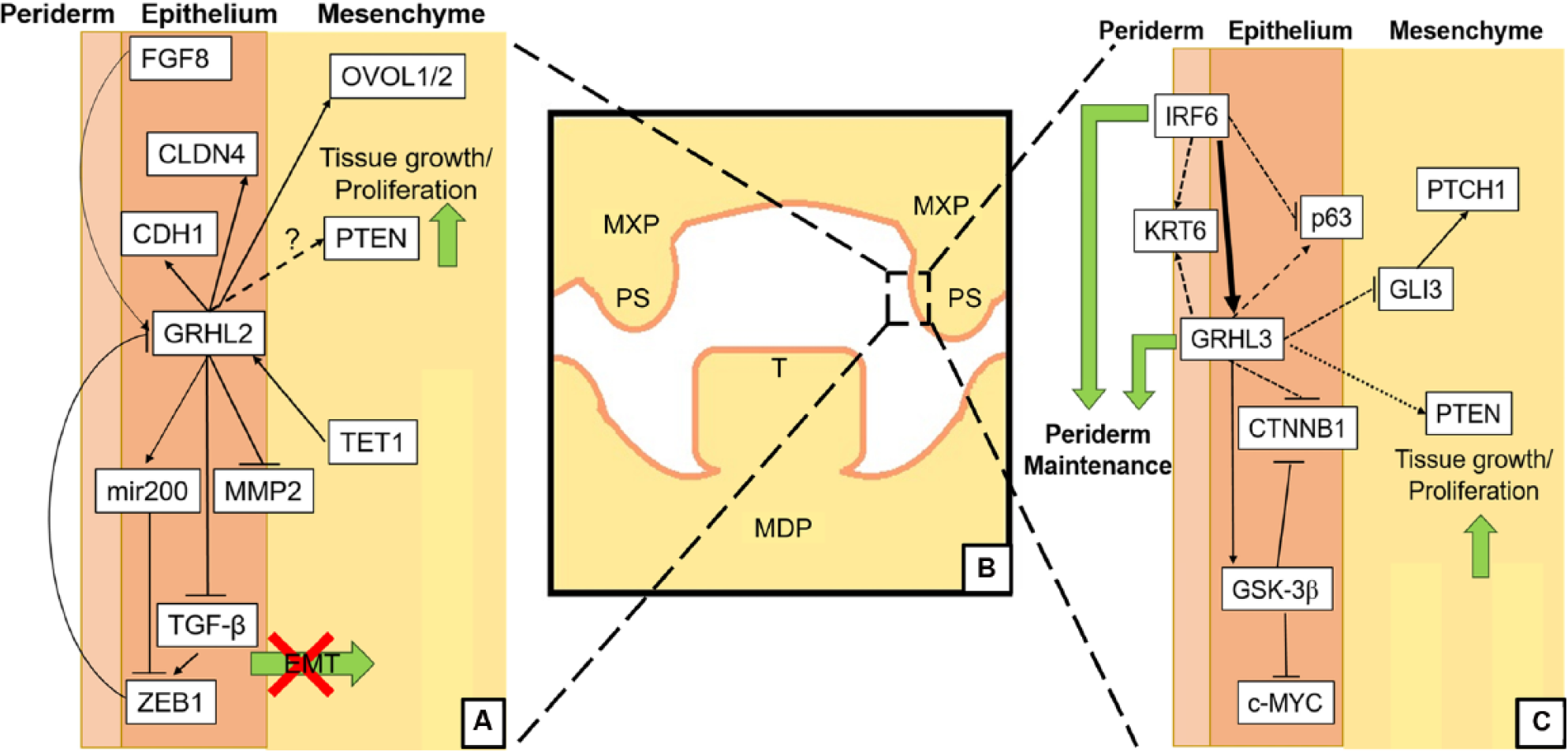
Putative Grhl2/Grhl3 signaling pathways in palatogenesis. (
Grhl3 may regulate many of the same genes as Grhl2 within the palate (Fig. 4). We have shown that Grhl3 directly transactivates Gsk3β to suppresses c-myc signaling in oral epithelial cancer cells (Georgy et al. 2015), and since Gsk3β deficiency also leads to palatal clefting (Liu et al. 2007; He et al. 2010), Grhl3 could regulate palatal shelf epithelial fidelity via this pathway. In addition, Grhl3 regulates phosphatase and tensin homolog (PTEN) in squamous cell carcinoma of the skin (Darido et al. 2011), and phosphorylated PTEN is expressed throughout the developing oral epithelium (Cho et al. 2008), suggestive of possible cooperative regulation in the maintenance of oral epithelial fidelity. Last, Grhl3 has also been shown to regulate Ctnnb1 and Gli3 within the developing circumvellate papillae epithelium (Adhikari et al. 2017), indicating that Grhl3-dependent regulation of genes within the Shh pathway, which itself is a critical regulator of maxillofacial development, may underpin craniofacial formation.
Summary
grh/Grhl transcription factors play essential roles in the craniofacial development of flies, fish, mice, and humans. In mammals, Grhl2 and Grhl3 have different roles in palate formation. Grhl2 is required for outgrowth of the maxillary processes while Grhl3 is required for periderm differentiation, although both are required for maintenance of the oral epithelium. The latter explains why patients with GRHL3 mutations have CPO but does not fully explain why these patients sometimes have CL, CL/P, or lip pits. Although the discovery of a common regulatory network involving IRF6 and Grhl3 is an exciting advance, further detailed characterization of the earlier stages of palate development in Grhl3 and Irf6 nullizygous mouse models is required to determine specific functional interaction, particularly to determine the precise spatiotemporal expression of these genes. Further characterization of the myriad developmental pathways operating in craniofacial development is needed to fully elucidate the Grhl-dependent transcriptional network within the palatal epithelia.
Author Contributions
M.R. Carpinelli, M.E. de Vries, S. Dworkin, contributed to data interpretation, drafted and critically revised the manuscript; S.M. Jane, contributed to data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study was funded by the Department of Health, Australian Government, National Health and Medical Research Council grant APP1063837.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.