
Abstract
Results from microbiome studies on oral cancer have been inconsistent, probably because they focused on compositional analysis, which does not account for functional redundancy among oral bacteria. Based on functional prediction, a recent study revealed enrichment of inflammatory bacterial attributes in oral squamous cell carcinoma (OSCC). Given the high relevance of this finding to carcinogenesis, we aimed here to corroborate them in a case-control study involving 25 OSCC cases and 27 fibroepithelial polyp (FEP) controls from Sri Lanka. DNA extracted from fresh biopsies was sequenced for the V1 to V3 region with Illumina’s 2 × 300–bp chemistry. High-quality nonchimeric merged reads were classified to the species level with a prioritized BLASTN-based algorithm. Downstream compositional analysis was performed with QIIME (Quantitative Insights into Microbial Ecology) and linear discriminant analysis effect size, while PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) was utilized for bacteriome functional prediction. The OSCC tissues tended to have lower species richness and diversity. Genera Capnocytophaga, Pseudomonas, and Atopobium were overrepresented in OSCC, while Lautropia, Staphylococcus, and Propionibacterium were the most abundant in FEP. At the species level, Campylobacter concisus, Prevotella salivae, Prevotella loeschii, and Fusobacterium oral taxon 204 were enriched in OSCC, while Streptococcus mitis, Streptococcus oral taxon 070, Lautropia mirabilis, and Rothia dentocariosa among others were more abundant in FEP. Functionally, proinflammatory bacterial attributes, including lipopolysaccharide biosynthesis and peptidases, were enriched in the OSCC tissues. Thus, while the results in terms of species composition significantly differed from the original study, they were consistent at the functional level, substantiating evidence for the inflammatory nature of the bacteriome associated with OSCC.
Introduction
Oral cancer, predominantly oral squamous cell carcinoma (OSCC), is the 14th-most prevalent malignancy worldwide, accounting for 300,373 new cases and 145,343 deaths annually (Ferlay et al. 2013) . In the West, 74% of OSCC cases are attributed to tobacco smoking and alcohol consumption (Petersen 2009), while in South Asia and the Pacific, tobacco chewing with or without areca (betel) nut is the major risk factor (Gupta and Johnson 2014). A variable fraction of OSCC cases is associated with HPV infection (Shaikh et al. 2015), while around 15% remains unexplained by any of the known risk factors. In addition, and despite advances in cancer treatment modalities, OSCC continues to have a poor prognosis, with 5-y survival rates <50% in much of the world (Sklenicka et al. 2010). These challenges have encouraged scientists to search for novel risk factors and prognosis modifiers. Particularly, there has been increasing interest in the role of the microbiome in oral carcinogenesis (Perera et al. 2016).
In the 1990s, the relationship between carcinogenesis and bacteria was first established by demonstrating the causative role of Helicobacter pylori in gastric cancer (Kim et al. 2011). Since then, tremendous efforts have been made to explore the relationship between bacteria and cancer in other sites of the body, including the oral cavity. Interest has even recently shifted from studying the role of a single species to that of the entire microbial community, driven by the emerging concept of microbial dysbiosis and its role in health and disease (Sheflin et al. 2014). The epidemiologic association between bacteria and OSCC was assessed in several studies employing technologies ranging from cultivation to close-ended molecular techniques (e.g., DNA-DNA hybridization) and next-generation sequencing of the 16S rRNA gene (microbiome analysis; Perera et al. 2016). However, the results from those studies were inconsistent regarding what species or microbial shifts are associated with OSCC. This is in part due to the methodological variations among those studies but, more important, because they were limited to compositional analysis, which does not account for functional redundancy in the human microbiome: the fact that unrelated species can have similar functions and thus replace one another in different ecosystems (e.g., samples from different subjects) without change in overall ecosystem functioning (Tian et al. 2017).
Thus, more consistent and perhaps more informative results are likely to be obtained with functional rather than compositional analysis. So far, functional analysis of the bacteriome within OSCC tissues is limited to a very recent study involving patients with OSCC from Yemen (Al-hebshi et al. 2017). In that study, a high-resolution taxonomy assignment algorithm was employed to characterize the composition of the bacteriome associated with OSCC to the species level, which identified several species, including Fusobacterium nucleatum and Pseudomonas aeruginosa, in association with OSCC. More important, the study performed functional prediction analysis, which revealed enrichment of proinflammatory bacterial attributes, such as lipopolysaccharide (LPS) synthesis, flagellar assembly, and bacterial chemotaxis in the tumor tissue. This is a very important finding given the well-established role of inflammation in cancer, but it has not been confirmed by another study.
The aim of this study was to corroborate the Yemeni study with a cohort of OSCC cases from Sri Lanka. Identical sequencing strategy and data analysis pipeline to those of the Yemeni study were employed; however, oral fibroepithelial polyp (FEP) tissues from broadly matched patients, rather than epithelial swabs from healthy individuals, were used as controls. FEP is a benign hyperplasia of oral mucosa that is treated by excision and thus represents a better control for within-tissue microbiome analysis.
Materials and Methods
Study Population and Sampling
The study subjects and sampling were previously described in detail (Perera et al. 2017). In brief, a subsample was included from a large-scale case-control study conducted in Sri Lanka. The study cohort comprised 25 Sinhala males with histologically confirmed OSCC involving the buccal mucosa or tongue (cases) and 27 Sinhala males with FEP from the same anatomic sites (controls). Appendix Table 1 shows the demographic and clinical characteristics of the study groups. This study was approved by the University of Peradeniya, Sri Lanka (FRC/FDS/UOP/E/2014/32), and the Griffith University Human Research Ethics Committee, Australia (DOH/18/14/HREC). Each participant provided a written consent. Deep tissue samples (~100 mg each) were dissected from fresh incisional biopsies, avoiding contamination from the tumor surface, and stored at −80 °C.
This reports conforms, where applicable, to the STROBE guidelines (Strengthening the Reporting of Observational Studies in Epidemiology).
DNA Extraction and 16S rRNA Sequencing
The methods for DNA extraction and sequencing were previously described (Perera et al. 2017). Briefly, DNA was extracted with the Gentra Puregene Tissue kit (Qiagen) following the manufacturer’s protocol for solid tissue with minor modifications. The V1 to V3 region of the 16S rRNA gene was amplified for sequencing with the degenerate primers 27FYM (5′-AGAGTTTGATCMTGGCTCAG-3′) and 519R (5′-GW ATTACCGCGGCKGCTG-3′). Library preparation, indexing, and sequencing were performed at the Australian Centre for Ecogenomics (University of Queensland, Australia) with the v3 2 × 300–bp chemistry on a MiSeq platform (Illumina).
Data Preprocessing
Raw data were deposited at Sequence Read Archive under project no. PRJNA415963. Preprocessing of data—including primer trimming, merging, quality filtration, alignment, and chimera check—were performed as detailed previously (Al-hebshi et al. 2017) with the exception of using a less stringent Q-score average cutoff for the sliding 50-nucleotide window (30 instead of 35). The high-quality nonchimeric merged reads were classified with the prioritized species-level taxonomy assignment algorithm (Al-hebshi et al. 2015) as implemented by Al-hebshi et al. (2017). In brief, each read was BLASTN searched against 4 databases of 16S rRNA gene reference sequences at alignment coverage and percentage identity ≥98% and then assigned species-level taxonomy of the hit with highest percentage identity and bit score, and belonging to the highest-priority reference set. Reads with no matches at the set cutoffs were subject to de novo operational taxonomic unit calling and assigned to the closest species.
Compositional Analysis and Functional Prediction
Compositional analysis and functional prediction were performed as previously described (Al-hebshi et al. 2017). In brief, QIIME (Quantitative Insights into Microbial Ecology; Caporaso et al. 2010) was used for generation of taxonomy plots/tables and rarefaction curves; calculation of species richness, coverage, and diversity; and clustering of samples with principal coordinate analysis (PCoA). The microbial metagenomes were imputed from the 16S rRNA data with PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) based on KEGG Orthology genes and pathways (Langille et al. 2013). Differentially abundant taxa, genes, and pathways between the cases and controls were sought with linear discriminant analysis effect size (LEfSe; Segata et al. 2011).
Results
Sequencing and Data Processing Statistics
The sequencing run generated 3,277,451 raw paired reads. Nearly 30% of them had primer mismatches; 35% could not be successfully stitched with PEAR; and 15.6% did not pass quality filtration. An additional 157,601 reads were filtered out in the alignment and chimera check steps, leaving a final of 451,048 merged reads with an average length of 482 bp. Four samples had read counts <3,000 and were thus excluded from further analysis. Of the remaining reads, 89% were successfully classified to the species level; the rest were identified as additional chimeras, did not return BLASTN matches, or formed singleton operational taxonomic units. The number of classified reads averaged 8,227 ± 2,923 per subject (range, 4,254 to 18,988 reads). The results described here were based on a minimum read count per species of 1. Results for higher cutoffs (10 and 100) can be found at http://bioinformatics.forsyth.org/ftp/publication_data/20161213/.
Bacteriome Compositional Profile
A total of 1,072 species-level taxa (including 373 potentially novel species) belonging to 272 genera and 19 phyla were detected in the 48 samples. The per-sample and per-group relative abundances and detection frequencies of taxa at each level are presented in Appendix Data Sets 1 to 3.
Figure 1 illustrates the distribution of top 5 phyla, top 15 genera, and top 25 species detected in the samples. Phyla Fusobacteria, Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria accounted for the majority of the sequences. At the genus level, Streptococcus, Rothia, Leptotrichia, Gemella, Capnocytophaga, Fusobacterium, Prevotella, Haemophilus, and Granulicatella accounted for the majority of the bacteriome in most samples. Pseudomonas, Citrobacter, Klebsiella, and Entrobacter were highly abundant in a few samples, which inflated their average abundance. At the species level, there was significant intersample variation, but Rothia mucilaginosa, Gemella haemolysans, Streptococcus mitis, F. nucleatum subsp. polymorphum, Streptococcus sp. oral taxon 431, Streptococcus dysgalactiae, Haemophilus parainfluenzae, Capnocytophaga sputigena, Capnocytophaga leadbetteri, and Prevotella melaninogenica were the most abundant on average: Citrobacter koseri dominated in 3 samples, which skewed its average abundance.
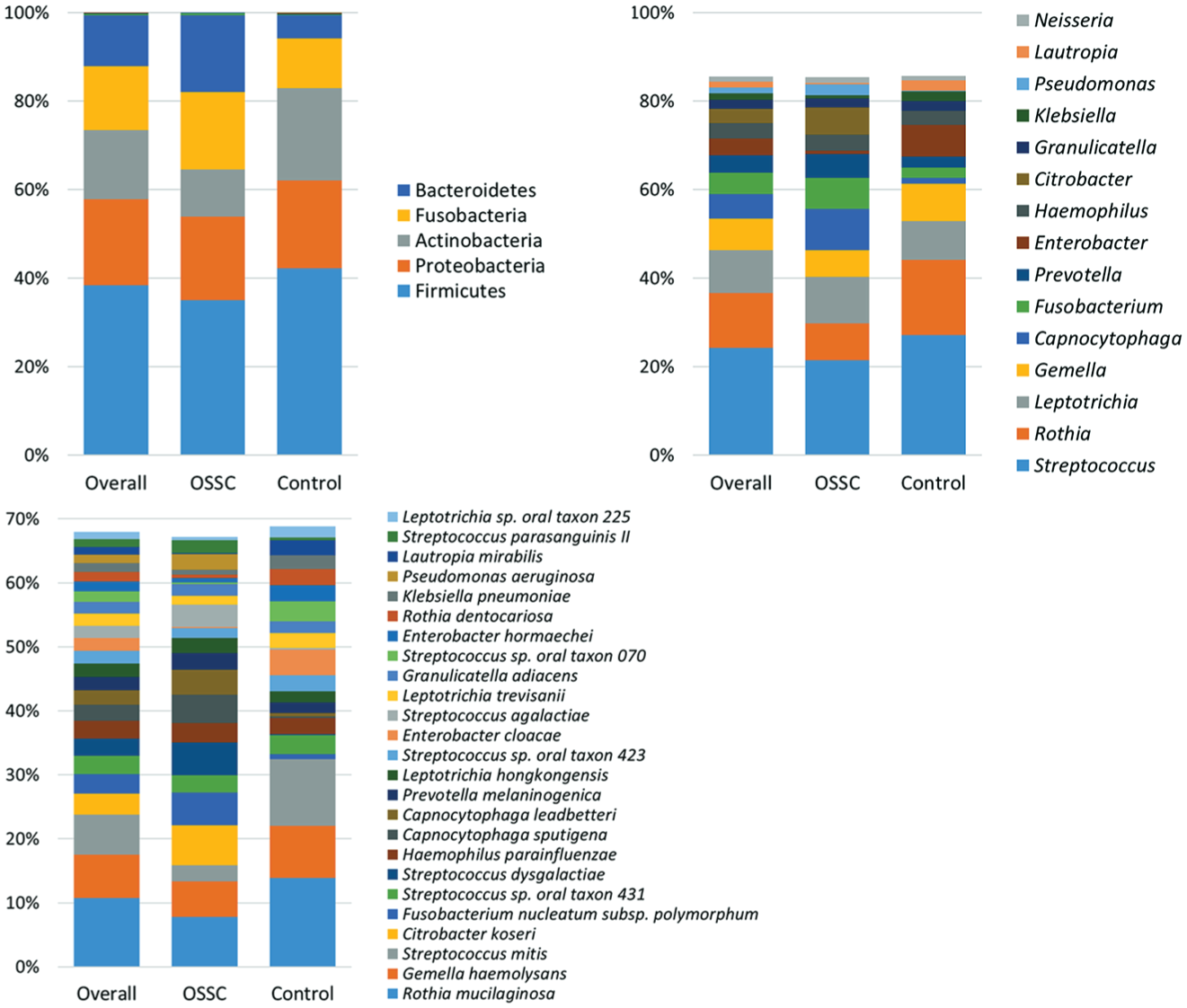
Relative abundances of the top 5 phyla, top 15 genera, and top 25 species identified. OSCC, oral squamous cell carcinoma.
OSCC versus FEP: Composition
The number of species detected in the cases and controls was 688 and 810, respectively, with 426 species in common. The number of species varied from 20 to 183 in the OSCC group and 23 to 229 in the FEP control group (Appendix Data Set 3). Species richness and α-diversity were higher in the controls as compared with the cases, but the differences were not statistically different (Table; Appendix Fig.). In PCoA, the cases and controls tended to form separate clusters based on binary data (unweighted UniFrac) and abundance data (weighted UniFrac; Fig. 2), with the clusters being statistically different as assessed by analysis of similarities (P = 0.01 and 0.03, respectively).
Species Richness, α-diversity, and Coverage Calculated from the Rarefied BIOM.

Values are presented as mean ± SE. Differences statistically not significant (P > 0.05), Mann-Whitney test.
BIOM, Biological Observation Matrix; FEP, fibroepithelial polyp; OSCC, oral squamous cell carcinoma.
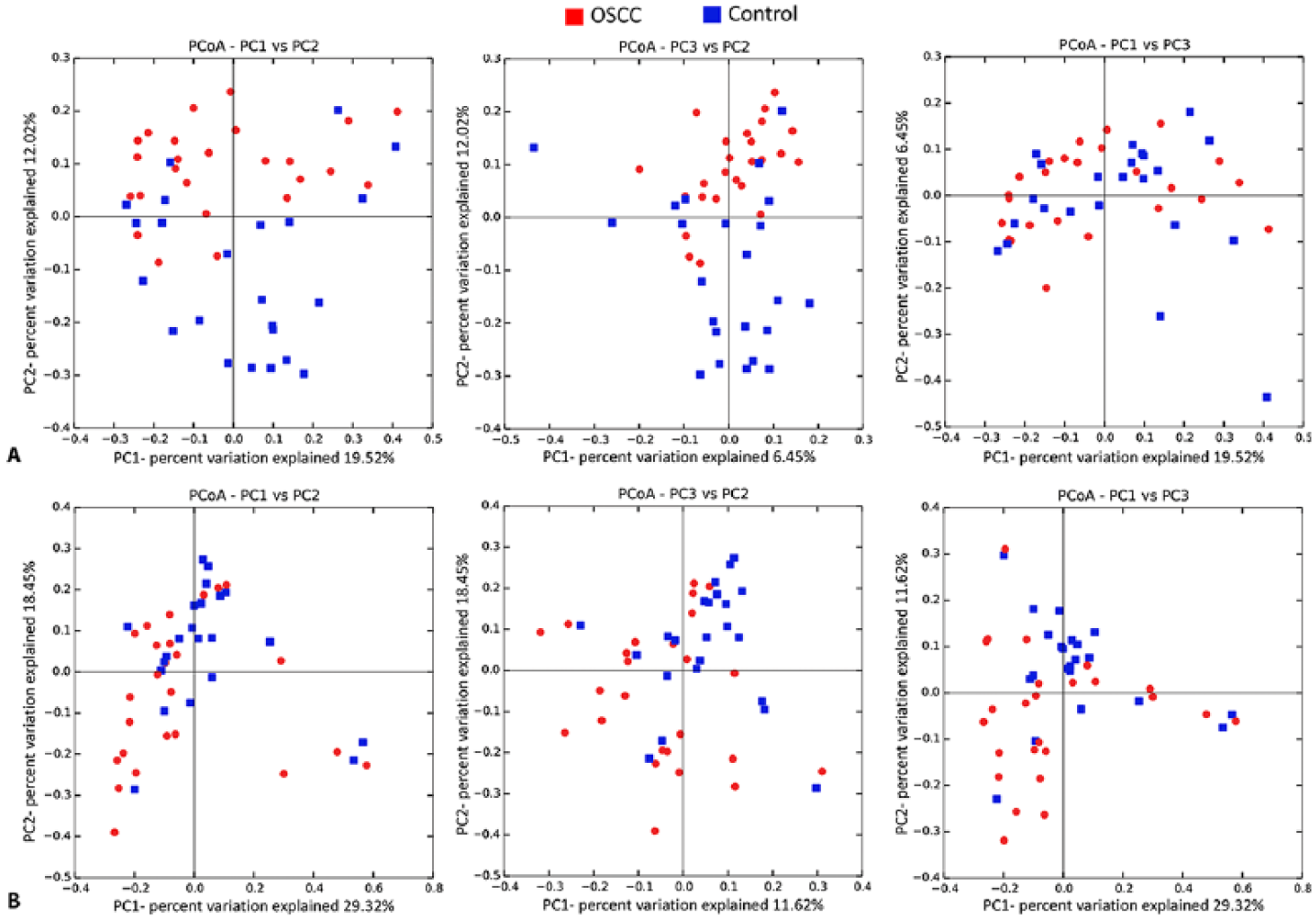
β-diversity analysis. Clusters formed by principle coordinate analysis (PCoA) based on (
The differentially abundant genera and species between the cases and controls are displayed in Figure 3. Genera Capnocytophaga, Pseudomonas, and Atopobium were associated with OSCC, while Lautropia, Staphylococcus, Propionibacterium, and Sphingomonas were the most significantly abundant in FEP. At the species level, Campylobacter concisus, Prevotella salivae, Prevotella loeschii, and Fusobacterium oral taxon 204 were enriched in the cases, while 7 Streptococcus species, 2 Rothia species, Lautropia mirabilis, and Leptotrichia oral taxon 225, among others, were significantly overrepresented in the controls. Using G-test instead of LEfSe to explore differentially abundant taxa identified additional associations. Among others, F. nucleatum subsp. polymorphum, S. dysgalactiae, C. koseri, and P. aeruginosa were significantly more abundant in OSCC, while S. mitis and Staphylococcus epidermidis was enriched in FEP (Appendix Table 2).
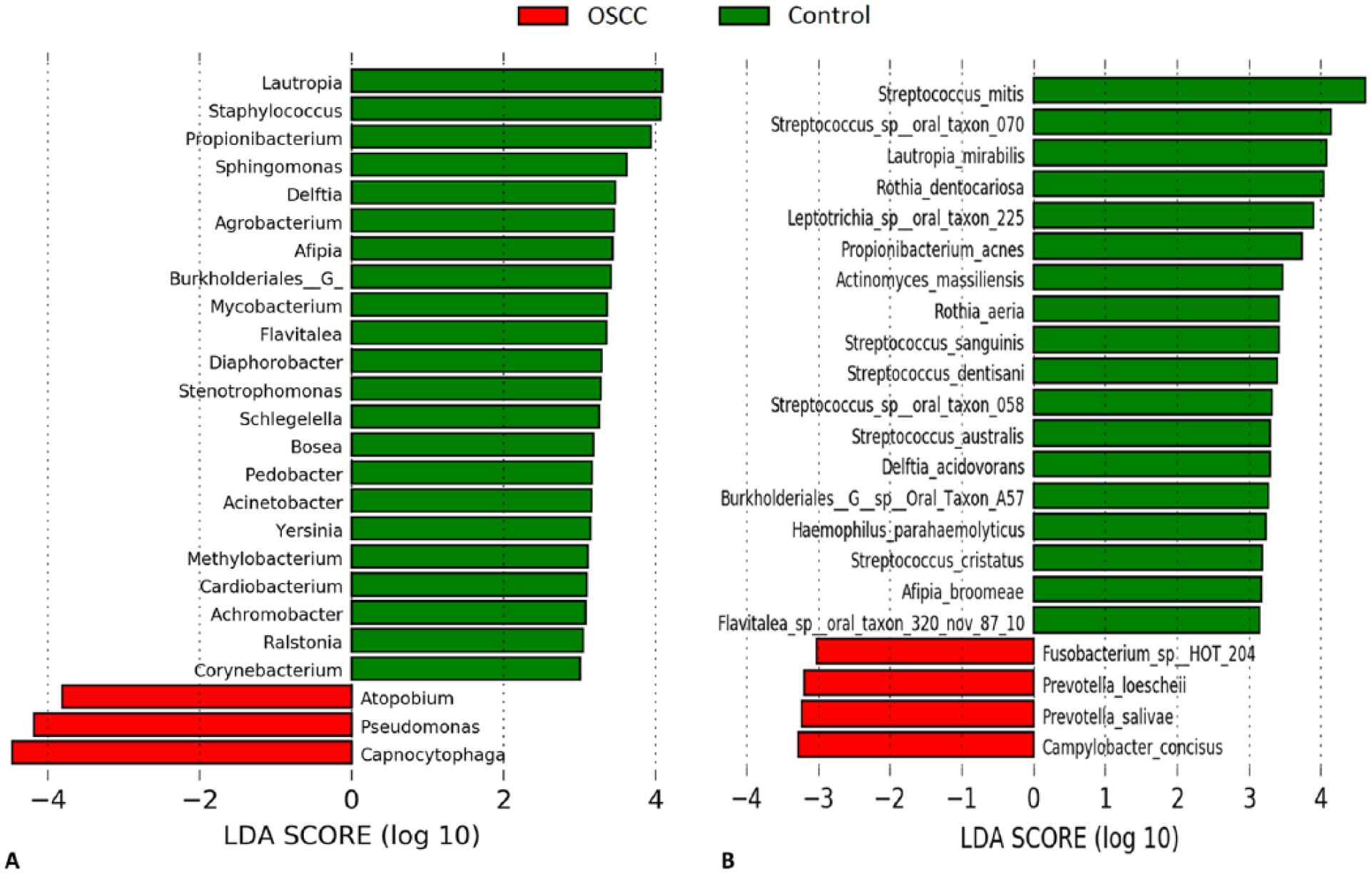
Differentially abundant taxa. (
OSCC versus FEP: Functional Potential
Figure 4 shows the microbial genes and pathways enriched in each study group. Genes encoding for a variety of enzymes, such as transketolase, pyruvate formate lyase activating enzyme, formate C-acetyltransferase, aspartokinase/homoserine dehydrogenase and nitroreductase/dihydropteridine reductase, were significantly more abundant in the OSCC cases. In contrast, genes responsible for production of iron complex transport system proteins, aspartyl-tRNA(Asn)/glutamyl-tRNA(Gln) amidotransferase subunits A and B, among others, were overrepresented in FEP. At the pathway level, LPS biosynthesis, energy metabolism, membrane and intracellular structural molecules, and peptidases were enriched in OSCC tissues, whereas valine, leucine and isoleucine biosynthesis, glycolysis/glucogenesis, base excision repair, and protein kinases, among others, were more abundant in FEP tissues.
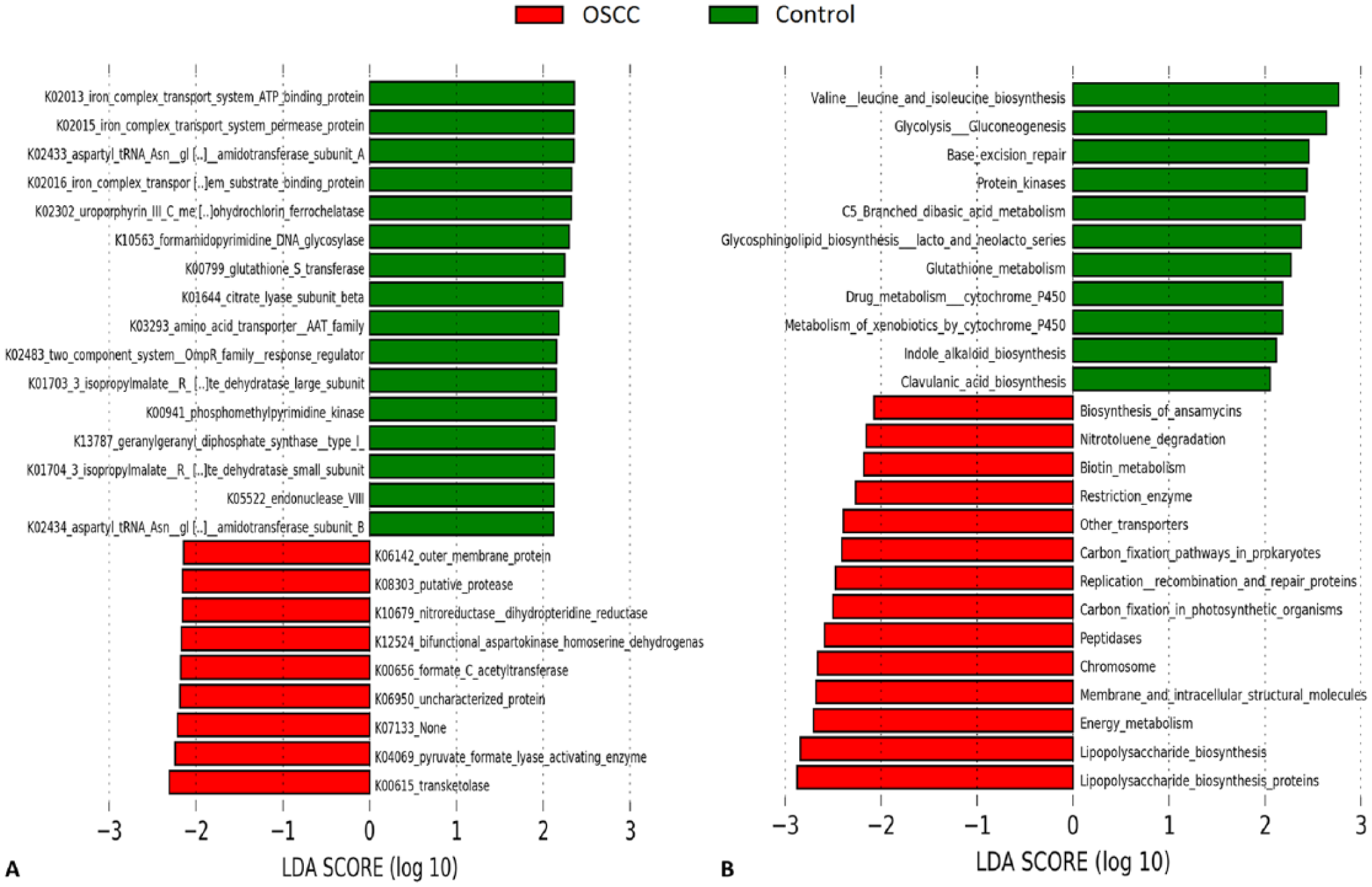
Differentially enriched functions. Microbial (
Discussion
Microbial dysbiosis closely tied to host inflammation has been demonstrated to play a role in the etiology of colon, gastric, esophageal, pancreatic, breast, and gall bladder carcinomas (Sheflin et al. 2014). In line with this, Al-hebshi et al. (2017) reported presence of a dysbiotic proinflammatory bacteriome within tissues of OSCC from a Yemeni patient cohort. In this case-control study, we demonstrate comparable findings in a Sri Lankan cohort as in the Yemeni study after following a similar approach: deep tumor tissue samples rather than surface swabs were analyzed; the V1 to V3 region, amplified with the same primers, was sequenced; and the same species-level taxonomy assignment and functional prediction algorithms were employed. OSCC is highly prevalent in Yemen and Sri Lanka and is mainly attributed to use of smokeless tobacco products in both countries, usually involving areca nut as well in the latter (Gupta and Johnson 2014; Nasher et al. 2014). Nevertheless, a number of methodological differences between the 2 studies are to be noted that may account for some of the variation in the results. In the study by Al-hebshi et al. (2017), deep epithelial swabs from healthy individuals were used as control samples. In contrast, the current study used deep FEP tissues instead. FEP is a common reactive hyperplasia of the oral mucosa, with a core of vascular connective tissue covered by basically normal stratified squamous epithelium, with no evidence of dysplasia or neoplastic changes. As we were investigating bacteria in deep cancerous tissue, we used controls also from deep tissues of benign oral mucosal lesions; surface swabs would have been contaminated by salivary flora and would not represent the microenvironment of deep tissues. Another methodological difference between the studies is that while the current study included only tongue and buccal cancer, that by Al-hebshi et al. also involved tumors from the floor of the mouth and the gum. Finally, we employed a different DNA extraction protocol.
The patients with cancer were older than the controls—an inevitability given that OSCC is usually diagnosed at an older age than is FEP. They also had more severe periodontal disease, partly because they were older. More apposite is that they had a higher exposure to risk factors common to oral cancer and destructive periodontitis: tobacco and betel nut consumption, poor diet, and poor oral hygiene . These factors may have driven some of the differences in the microbiome between the groups. However, none of the classical periodontal pathogens, such as Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola, was enriched in the OSCC tissues.
Although not statistically significant, the Sri Lankan tumors tended to have lower species richness and diversity than the controls, which is not consistent with the results by Al-hebshi et al. (2017), in which the OSCC and control samples displayed similar bacterial species richness and diversity. This is certainly not due to differences in nature of the controls between the studies, because a direct comparison shows that richness and diversity were higher in the tumors from the Yemeni cohort (122.2 ± 49.9 vs. 82.9 ± 37.8): this is probably due to the differences in final sequencing depths between the studies (Yemeni: 14,357 ± 4,499 vs. Sri Lanka: 8,227 ± 2,923 reads per subject).
Genera Capnocytophaga, Pseudomonas, and Atopobium showed association with OSCC. This is consistent with previous studies with respect to Capnocytophaga (Mager et al. 2005; Hooper et al. 2007; Zhao et al. 2017) and Pseudomonas (Al-hebshi et al. 2017). The association of Atopobium with OSCC is reported here for the first time; interestingly, this genus was recently identified in association with chronic periodontitis (Ai et al. 2017). At the species level, LEfSe analysis revealed association of a different panel of species with OSCC in the Sri Lankan cohort as compared with that reported by Al-hebshi et al. (2017). Unlike in the current study, no Prevotella species were enriched in the tumors from the Yemeni cohort. However, C. concisus and Fusobacterium oral taxon 204, which were enriched in this study, may be functional equivalents to the Campylobacter oral taxon 44 and F. nucleatum, respectively, found in association with OSCC in the Yemeni samples. Furthermore, the G-test identified F. nucleatum subsp. polymorphum as well as P. aeruginosa to be associated with OSCC in the current cohort, although the strength of association was lower than reported by Al-hebshi et al. (2017). There is substantial evidence for the carcinogenicity of F. nucleatum from in vitro and animal studies (Uitto et al. 2005; Binder Gallimidi et al. 2015); it has also been implicated in colorectal carcinoma (Kostic et al. 2012). P. aeruginosa has not been so far linked to cancer; however, it possesses virulence factors, such as LPS, flagella, and exotoxin U, with demonstrated hyperinflammatory properties that may play a role in carcinogenesis (de Lima et al. 2012; Gellatly and Hancock 2013).
The genera associated with FEP, with the exception of Lautropia, were very different from those found to be enriched in normal buccal epithelium of the Yemeni cohort, which is not surprising because the samples are different in nature. It may be that some of the taxa found to be associated with FEP, such as Staphylococcus, Propionibacterium, and Sphingomonas, play a role in its etiology, a possibility that warrants further investigation. At the species level, however, the results were largely consistent with those reported by Al-hebshi et al. (2017). Most of the streptococci found to be overrepresented in the FEP samples from the Sri Lankan cohort, particularly S. mitis, were also enriched in the normal buccal epithelium samples from the Yemeni cohort; Rothia spp. (though not the same species) and L. mirabilis were also enriched in both groups. Schmidt et al. (2014) reported similar findings for Streptococcus and Rothia, although it is important to note that their control samples were from contralateral, clinically normal mucosal sites. Because of the phenomenon of field cancerization—which recognizes that adjacent tissues other than the primary site of the tumor (the whole mouth here) have been exposed to topical carcinogens and can thus undergo malignant transformation (Mohan and Jagannathan 2014)—we used control samples from ethnically matched subjects rather than opposite anatomic sites in the mouth of the OSCC subjects. The ability of the host to resist or be susceptible to particular carcinogenic challenges also has systemic drivers, so tissues elsewhere in the same individual are inappropriate controls.
Unlike the results of compositional analysis, the results from functional prediction analysis were consistent between our studies. Particularly, LPS biosynthesis pathways were enriched in both cohorts. LPS are potent inflammatory molecules with cancer-promoting properties in vivo and were shown to enhance invasion in pancreatic cancer via the TLR/MyD88/NF- NF-κB pathway (Ikebe et al. 2009). They can also facilitate breast cancer and colorectal carcinoma metastasis by activation of the prostaglandin E2-EP2 pathway (Li et al. 2015) and stimulation of toll-like receptor TLR4 (Hsu et al. 2011), respectively. Apart from LPS, peptidases were overrepresented in OSCC. There is some evidence to suggest that bacterial proteases play a role in inflammatory disorders such as inflammatory bowel disease (Carroll and Maharshak 2013); in fact, a cysteine protease released by the oral pathogen Porphyromonas gingivalis was shown to upregulate production of proinflammatory cytokines (Giacaman et al. 2009). This study, thus, is in line with the study by Al-hebshi et al. (2017) in that the bacteriome found within tissues of OSCC is functionally proinflammatory, which is a highly relevant finding given the established role of inflammation in cancer. Notably, a very recent microbiome study of OSCC also performed functional prediction analysis but did not identify any bacterial inflammatory attributes in association with the tumors (Zhao et al. 2017). However, the study involved analysis of surface swabs rather than biopsies, indicating that the inflammatory bacteriome is enriched only in the body of the tumor.
It should be emphasized that the bacteriome associated with OSCC may be a consequence of the tumor microenvironment, rather than a proximate etiologic factor. A plethora of factors in the microenvironment, such as nutrient availability, pH, attachment ligands, and immune elements, likely shape the composition and function of the microbial community within the tumor tissue. Moreover, lifestyle-related risk habits of the cases, such as use of masticatory substances, poor oral hygiene status, and periodontal disease status, may influence oral bacterial colonization in OSCC tissues. That, however, does not mean that the microbial community does not in turn modify the behavior and progression of the tumor.
In conclusion, this case-control study confirms that a dysbiotic, inflammatory bacteriome is associated with OSCC and that microbial communities with different species composition can be functionally similar: an indirect evidence of functional redundancy among oral bacteria taxa. This study generates hypotheses that need to be confirmed in direct functional analysis with a metatranscriptomic approach.
Author Contributions
M. Perera, contributed to conception, design, data acquisition and interpretation, critically revised the manuscript; N.N. Al-hebshi, contributed to design, data analysis, and interpretation, drafted the manuscript; I. Perera, contributed to design, data acquisition, and interpretation, drafted the manuscript; D. Ipe, contributed to data acquisition, critically revised the manuscript; G.C. Ulett, contributed to design, critically revised the manuscript; D.J. Speicher, contributed to data acquisition and interpretation, critically revised the manuscript; T. Chen, contributed to data analysis, critically revised the manuscript; N.W. Johnson, contributed to conception, design, and data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
DS_10.1177_0022034518767118 – Supplemental material for Inflammatory Bacteriome and Oral Squamous Cell Carcinoma
Supplemental material, DS_10.1177_0022034518767118 for Inflammatory Bacteriome and Oral Squamous Cell Carcinoma by M. Perera, N.N. Al-hebshi, I. Perera, D. Ipe, G.C. Ulett, D.J. Speicher, T. Chen and N.W. Johnson in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Field-based data and sample collection was privately funded by M.P. and I.P. M.P. received Griffith University Higher Degree Scholarships for International Students.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.