
Abstract
Tooth eruption is a complex process requiring precise interaction between teeth and adjacent tissues. Molecular analysis demonstrates that bone remodeling plays an essential role during eruption, suggesting that a parathyroid hormone 1 receptor (PTH1R) gene mutation is associated with disturbances in bone remodeling and results in primary failure of eruption (PFE). Recent research reveals the function of PTH1R signaling in mesenchymal progenitors, whereas the function of PTH1R in mesenchymal stem cells during tooth eruption remains incompletely understood. We investigated the specific role of PTH1R in Prx1+ progenitor expression during eruption. We found that Prx1+-progenitors occur in mesenchymal stem cells residing in alveolar bone marrow surrounding incisors, at the base of molars and in the dental follicle and pulp of incisors. Mice with conditional deletion of PTH1R using the Prx1 promoter exhibited arrested mandibular incisor eruption and delayed molar eruption. Micro–computed tomography, histomorphometry, and molecular analyses revealed that mutant mice had significantly reduced alveolar bone formation concomitant with downregulated gene expression of key regulators of osteogenesis in PTH1R-deficient cells. Moreover, culturing orofacial bone-marrow-derived mesenchymal stem cells (OMSCs) from Prx1Cre;PTH1Rfl/fl mice or from transfecting Cre recombinase adenovirus in OMSCs from PTH1Rfl/fl mice suggested that lack of Pth1r expression inhibited osteogenic differentiation in vitro. However, bone resorption was not affected by PTH1R ablation, indicating the observed reduced alveolar bone volume was mainly due to impaired bone formation. Furthermore, we found irregular periodontal ligaments and reduced Periostin expression in mutant incisors, implying loss of PTH1R results in aberrant differentiation of periodontal ligament cells. Collectively, these data suggest that PTH1R signaling in Prx1+ progenitors plays a critical role in alveolar bone formation and periodontal ligament development during eruption. These findings have implications for our understanding of the physiologic and pathologic function of PTH1R signaling in tooth eruption and the progression of PFE.
Keywords
Introduction
Parathyroid hormone 1 receptor (PTH1R) is the receptor for parathyroid hormone (PTH) and parathyroid hormone related peptide (PTHrP). It has a profound effect on skeletal development and homeostasis (Juppner et al. 1991). Autosomal dominant or recessive mutations of PTH1R are associated with various clinical disorders. Homozygous mutations are responsible for Blomstrand type chondrodysplasia and Eiken syndrome. Heterozygous mutations result in Ollier’s disease and primary failure of eruption (PFE) (Jobert et al. 1998; Decker et al. 2008). PFE is a rare, nonsyndromic disorder solely found in dentition. It is characterized by incomplete tooth eruption despite clearance by bone resorption in the eruption path (Proffit and Vig 1981; Hanisch et al. 2018). When the eruption process halts before or during puberty, the development of adjacent alveolar bone is also influenced, leading to subemergence of affected teeth. Unlike patients with other eruption disorders, PFE patients often receive no beneficial prognosis from orthodontic extrusion (Frazier-Bowers et al. 2010). Thus, it is important to understand how PTH1R mutations cause PFE, which may unravel the etiology of PFE and improve early corrective diagnosis and treatment.
The correlation between PFE and PTH1R gene haploinsufficiency was discovered a decade ago. Continuing studies have confirmed that eruption disorders among PFE patients are linked to various PTH1R mutation sites (Decker et al. 2008; Grippaudo et al. 2018). However, the underlying mechanism by which PTH1R mutations lead to PFE is not fully understood. Animal studies demonstrated that a PTH1R ligand, PTHrP, plays an essential role during the eruption process. PTHrP-null mice experience early lethality, but restoring PTHrP expression in chondrocytes produces viable mice exhibiting tooth eruption and root formation failures. This phenotype was rescued by overexpressing PTHrP in epithelial cells (Philbrick 1998). Constitutive activation of PTH1R in osteocytes and odontoblasts with a collagenαI (ColI) promoter delayed tooth eruption (Tsutsui et al. 2008). Recent research has focused on PTH1R signaling in mesenchymal progenitors during the eruption process. Osx+ progenitors are in the dental follicle (DF) and root surface. Loss of PTH1R in Osx-expressing cells produced complete failure of tooth eruption with significantly truncated roots lacking periodontal ligaments (PDL) (Ono et al. 2016). Takahashi et al. established the PTHrP-creER mouse line and found PTHrP+ cells in molar DF. Ablation of PTH1R resulted in a cell fate shift of PTHrP+ cells to nonphysiologic cementoblast-like cells on the root surface, leading to a periodontal and root phenotype without any apparent alterations in osteoblasts or ankylosis (Takahashi et al. 2019). These findings emphasize the fundamental role of PTH1R in PDL development and tooth root formation during eruption (Richman 2019). However, imbalanced bone apposition and resorption are the putative factors underlying the progression of PFE (Grippaudo et al. 2018). The eruption path is cleared but there is no movement along this path in PFE, indicating a failure in the eruptive mechanism (Proffit and Vig 1981). The coordinated navigation of alveolar bone tightly controls the eruption process (Wise and King 2008). The mesenchymal progenitor populations relating to eruption and the function of PTH1R in stem cells responsible for alveolar bone development remain incompletely characterized. We thus undertook an analysis of mice in which PTH1R is conditionally ablated using the pair-related homeobox gene Prx1, a transcription factor that is highly expressed during limb bud formation and craniofacial development (Martin and Olson 2000). Prx1Cre targets early limb bud and craniofacial mesenchyme cells and is widely used for mesenchymal lineage-specific mutation models (Logan et al. 2002). Studies have used the Prx1Cre model to characterize specific gene functions in craniofacial mesenchyme (Wiszniak et al. 2016; Foster et al. 2017; Wang et al. 2018). Prx1-expressing cells and their postnatal progeny are characteristic of skeletal stem cells and play an essential role in skeletal development and bone homeostasis (Elefteriou and Yang 2011). Prx1Cre mice allow us to distinguish the role of PTH1R in osteogenesis from mesenchymal cells or bone deposition during eruption. Prx1+-progenitors identify mesenchymal stem cells in alveolar bone marrow and subsets of the dental follicle and pulp cells. Deletion of PTH1R in Prx1+-progenitors resulted in tooth eruption defects, possibly due to reduced alveolar bone growth and aberrant development of periodontal ligaments.
Materials and Methods
Mice
Floxed PTH1R and Prx1Cre mice were described previously (Kobayashi et al. 2002; Logan et al. 2002). B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J mice (Tmfl/fl) were purchased from Jackson Laboratory. Genomic DNA from tails was used for genotyping. All studies were approved by the Institutional Animal Care and Use Committee at the Harvard Medical School and State Key Laboratory of Oral Diseases, Sichuan University.
Detailed methods are in the Appendix.
Results
Distribution and Differentiation Potential of Prx1+ Progenitor Cells
Prx1 is expressed in the mesenchyme of tissues that develop into cartilage, bone, and tooth structures (Opstelten et al. 1991). We identified the pattern of Prx1-expressing cells during tooth eruption by crossing Prx1Cre and ROSA26tdTomato reporter mice in which Cre active cells express tdTomato. Analysis of Prx1Cre;Tmfl/+ mice at birth revealed that Prx1+ progenitor cells were predominantly found in alveolar bone surrounding incisors and at the base of molars. Prx1+ cells were detected in the dental follicle and dental pulp of incisors, but not in ameloblast lineage cells (Fig. 1A). At postnatal day 4 (P4), Prx1-expressing cells became preosteoblasts and osteoblasts as determined by immunostaining for Ostrix (Osx) and Osteopontin (Opn). Some Prx1+ lineage cells were observed in the dental pulp of molars (Fig. 1B, C). There were fewer Prx1+ cells in the maxilla than the mandible (Appendix Fig. 1D), consistent with previous findings (Fujimori et al. 2010). Immunostaining at P14 and P21 showed Prx1+ cells differentiating to Osx+ and Opn+ alveolar osteoblasts/osteocytes surrounding incisors and at the molar base, but not in molar cementum or PDL (Fig. 1D, E).
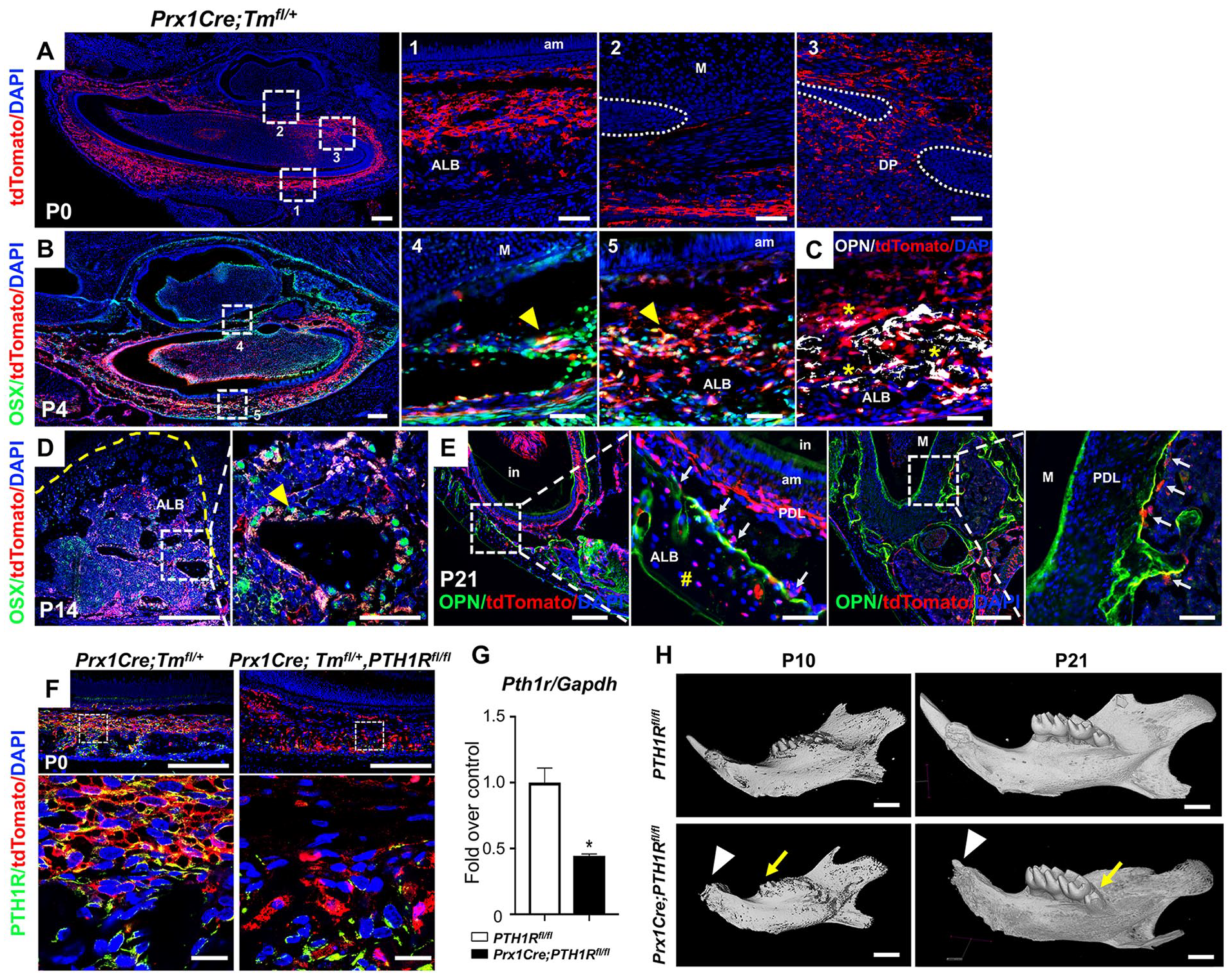
Loss of PTH1R in Prx1+ progenitors led to arrested tooth eruption. (
Conditional Deletion of PTH1R in Prx1+ Progenitors Led to Arrested Tooth Eruption
PTH1R is expressed in the mesenchyme of developing teeth (Liu et al. 1998). We have shown that Prx1Cre is present in dental mesenchyme, validating our mice with conditional deletion of PTH1R using the Prx1 promoter. This allows us to evaluate the role of PTH1R in dental mesenchyme during eruption. Prx1+-cells express PTH1R, which was significantly ablated in Prx1Cre;PTH1Rfl/fl (mutants) (Fig. 1F, G). A striking abnormality in mutants was mandibular incisor eruption failure. The eruption of mandibular incisors was not observed in mutants from P10 to P21 and after (Fig. 1H and Appendix Fig. 1A, B). Controls had complete eruption of mandibular and maxillary incisors at P21, while eruption was arrested in mutants. Upper incisor eruption was less affected, reflecting different Prx1Cre expression in the mandibular and maxilla regions. The arrested eruption of lower incisors resulted in an anterior open bite and overgrowth of the upper incisors in some mutants. This phenotype was observed in 6-mo-old mice, suggesting that the eruption of mutant mandibular incisors was arrested rather than delayed. HE staining at P0, P4, P10, and P14 showed that even though incisor eruption was disrupted in mutants, there were no differences in incisor cervical loop morphology (Appendix Fig. 2A). Amelogenin, Ameloblastin, and Enamelin gene expressions were comparable in mutant and control mandibles (Appendix Fig. 2B). We also observed defects in molar eruption. Comparison of molar eruption height above alveolar ridge by micro–computed tomography (µCT) scans revealed that mutants’ 1st molar eruption was delayed at P10. At P21, 1st and 2nd molars of mutants reached occlusion, showing comparable eruption height with controls, while the 3rd molar eruption height was significantly reduced (Appendix Fig. 1C). There were no root dilacerations, and root cementum was normal in mutant molars. Moreover, immunostaining showed unchanged CD200 expression between two groups surrounding molar root (Appendix Fig. 2C, D).
Prx1Cre;PTH1Rfl/fl Mice Exhibited Reduced Alveolar Bone Volume
We investigated the effects of diminished PTH1R signaling on tooth eruption by histology analysis. HE staining revealed that mutant mandibular bone had reduced alveolar bone volume at different time points throughout eruption (Fig. 2A). µCT analysis at P10 and P21 further suggested that mutants had reduced alveolar bone volume, whereas enamel and dentin volume of incisors remained unchanged (Fig. 2B–E). To assess bone apposition in mutants, histomorphometry was performed by calcein labeling. Bone formation parameters, including mineral surface/bone surface (MS/BS, %), mineral apposition rate (MAR, µm/d), and bone formation rate/bone surface (BFR/BS, µm3/µm2/d), were significantly decreased in Prx1Cre;PTH1Rfl/fl mice at P21 (Fig. 2F, G).
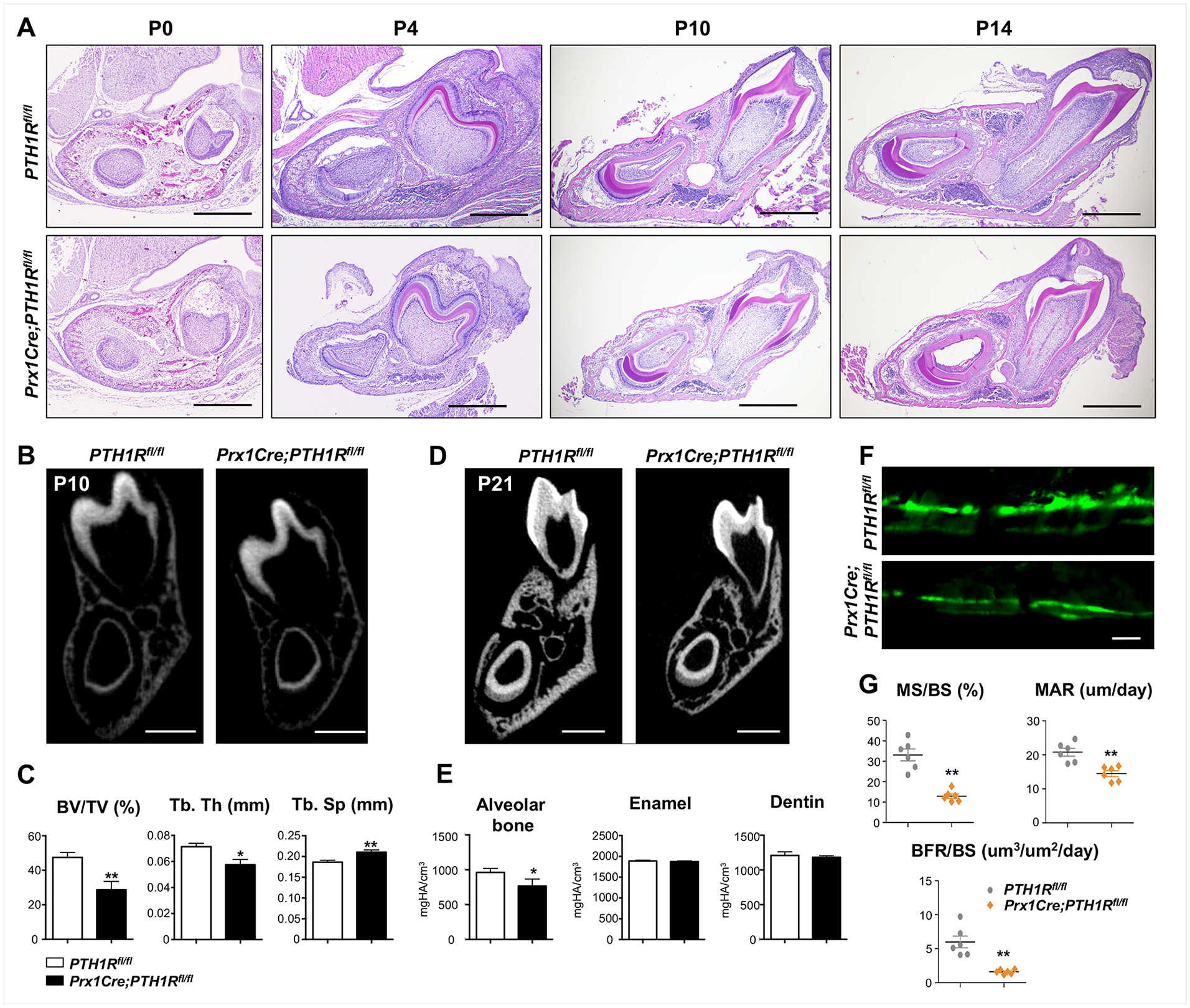
(
PTH1R-Deficient Progenitors Revealed Downregulated Osteogenesis
We investigated whether reduced alveolar bone volume in mutants was due to defects in formation or resorption by evaluating osteoblast and osteoclast function in mandibular bone. We determined which gene regulatory networks were affected by PTH1R deletion by using the transcriptomes of osteoblast-related markers. The results suggest that mutants had significantly reduced Runx2, Osx, Dentin matrix acidic phosphoprotein 1 (Dmp1), and Osteocalcin (Ocn) expression at P10 and P14 compared with controls (Fig. 3A). RUNX2 and OSX protein expressions were analyzed by immunofluorescent-double staining in Prx1Cre;Tmfl/+ and Prx1Cre;Tmfl/+,PTH1Rfl/fl mice where tdTomato marked Prx1+ lineage cells. Runx2+tdTomato+ and Osx+tdTomato+ cells were downregulated in mutants at several time points during eruption, indicating suppressed osteogenesis in PTH1R-ablated progenitor cells (Fig. 3B, C). Furthermore, immunofluorescent analyses of Col1 and tdTomato revealed that mutants had reduced Col1+ bone regions, whereas tdTomato+ osteoblast number/bone area were comparable with controls (Fig. 3D, E).
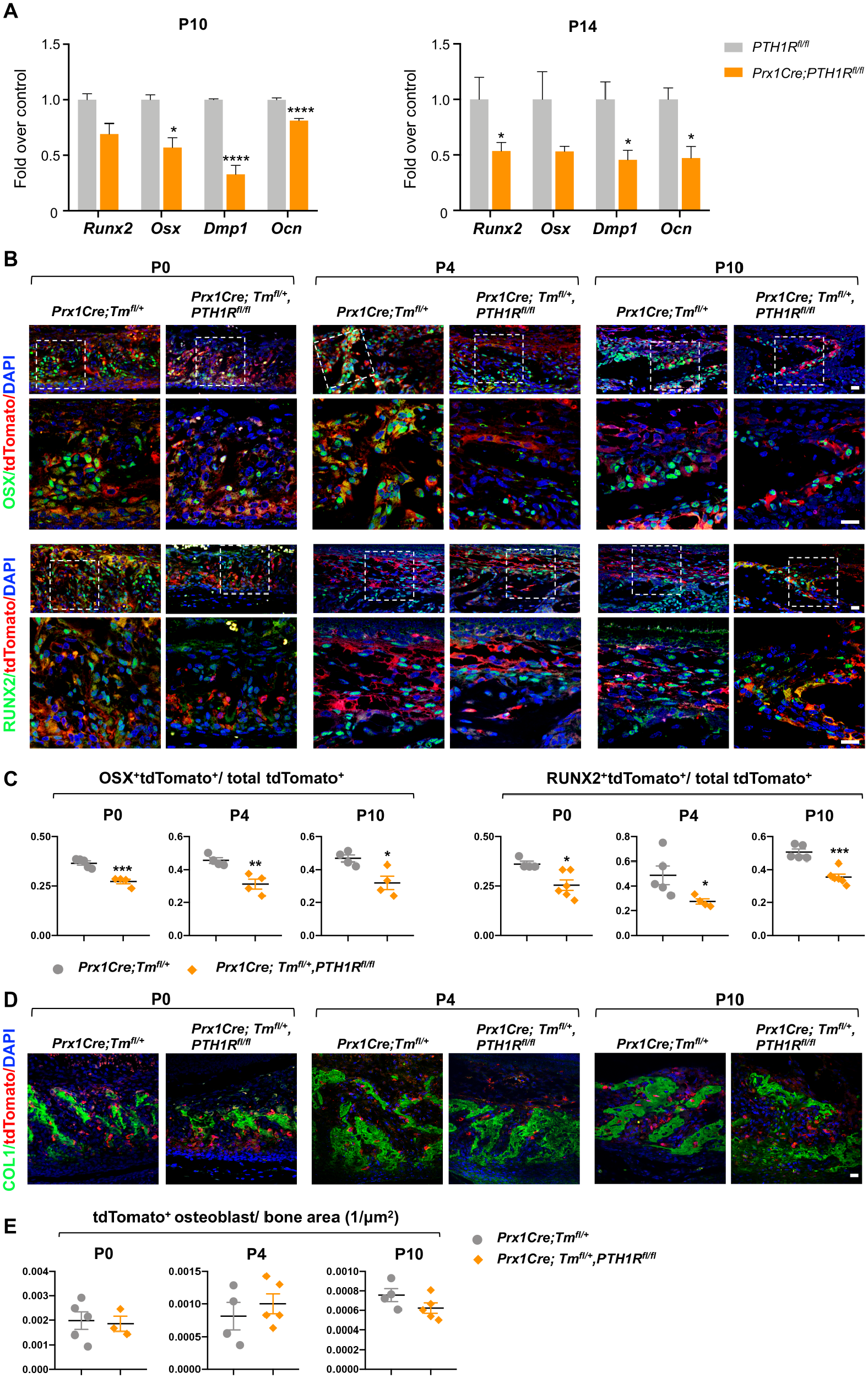
Prx1Cre;PTH1Rfl/fl mice revealed downregulated osteogenesis. (
Next, we investigated molecular correlates associated with reduced alveolar bone mass in Prx1Cre;PTH1Rfl/fl mice in vitro. We cultured orofacial bone-marrow-derived mesenchymal stem cells (OMSCs) derived from orofacial bone/bone-marrow (Yamaza et al. 2011). Flow-cytometric analysis of cell-surface markers showed that OMSCs were positive for CD90 and CD29 while negative for hematopoietic markers CD45 and CD11b (Fig. 4A). Moreover, OMSCs could further differentiate into osteogenic, adipogenic, and chondrogenic lineage cells, confirming their multilineage differentiation capacity (Appendix Fig. 3A–F). We then isolated OMSCs from PTH1Rfl/fl and Prx1Cre;PTH1Rfl/fl mice. After culture with osteogenic induction medium for 7 and 14 d, alkaline phosphatase (ALP) and alizarin red (ARS) staining intensities were markedly decreased in mutant OMSCs. Expression of osteogenic transcription factors and markers, including Osx, Runx2, Alp, Ocn, and Dmp1, were significantly reduced. These observations were confirmed by quantitative analyses of protein expression of RUNX2, OSX, and COLα1 (Fig. 4B–E). OMSCs from PTH1Rfl/fl mice also showed decreased mineralization in culture following treatment with CRE adenovirus compared with GFP adenovirus (Appendix Fig. 3G–I). Studies have shown important functions for various signaling pathways, such as BMP/TGF-β, Wnt/β-catenin in alveolar bone formation. Using qRT-PCR analysis, we found that expression of transcriptional targets of BMP/TGF-β such as Tgfbr1, Tgfb1, and Bmp1 were reduced in the mutant OMSCs, whereas downstream targets of Wnt/β-catenin signaling including Axin2, Ctgf, and Cyr61 remained unchanged (Fig. 4F). Several studies have shown that there is crosstalk between BMP/TGF-β signaling and PTH1R signaling. For instance, Tgfbr2 can directly phosphorylate PTH1R, which modulates PTH-induced endocytosis of both receptors and attenuates TGF-β and PTH1R signaling (Qiu et al. 2010). In our current analysis, the downregulation of the BMP/TGF-β pathway in PTH1R-deficient OMSCs supports the idea that these two pathways may interact in the osteogenic differentiation of OMSCs.
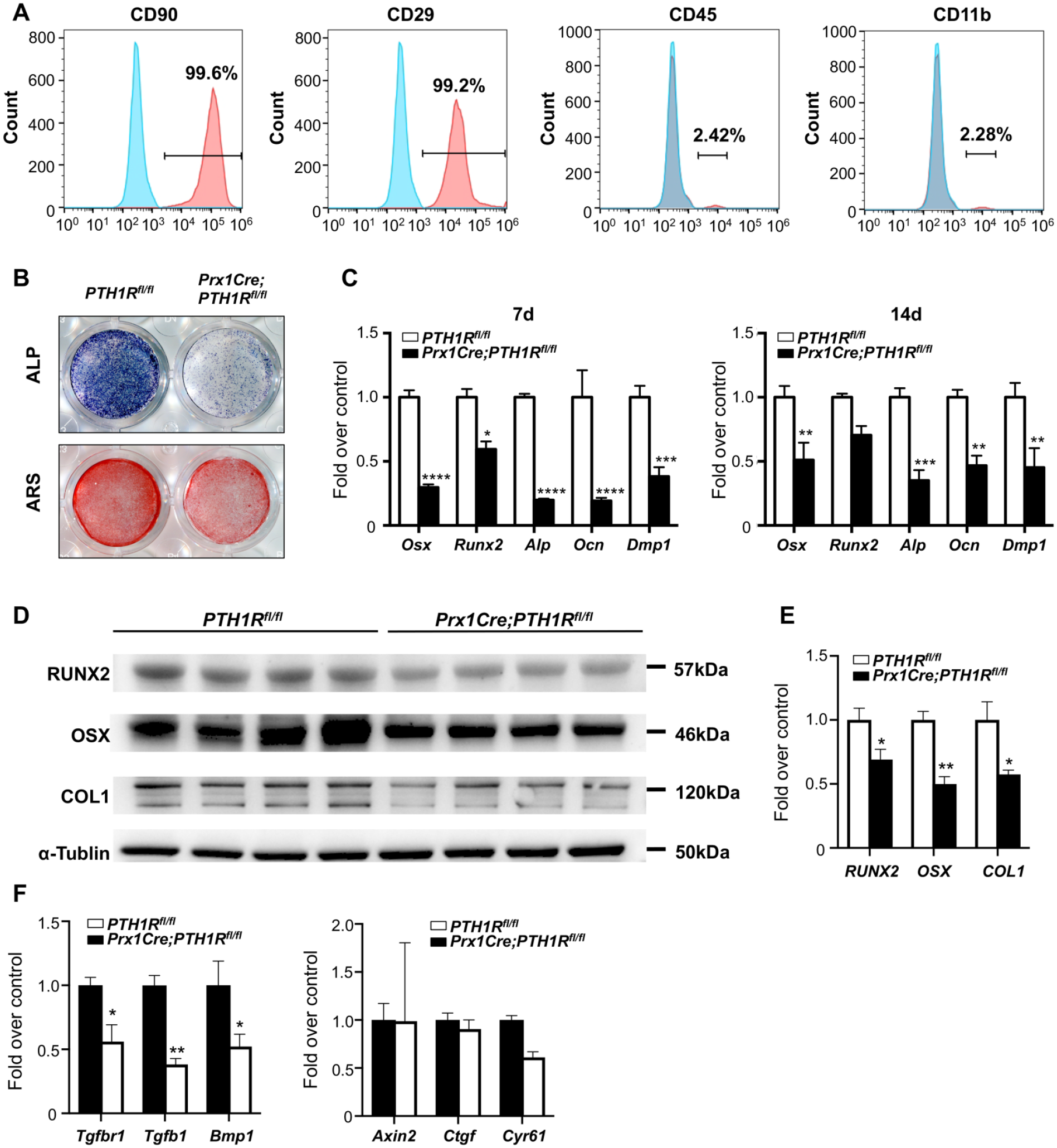
PTH1R-deficient OMSCs showed decreased osteogenic differentiation. (
Alveolar Bone Resorption Remained Unchanged with Diminished PTH1R Signaling
We investigated whether osteoclast alterations accounted for the phenotype. Osteoblast/osteoclast-relative gene expressions (i.e., Rankl, Opg, Rank) were measured in mandibles at P10 and P14. Consistent with TRAP staining, gene expression was unchanged between groups (Appendix Fig. 4A). TRAP+ cells were evident in mandibular alveolar bone surrounding molars and incisors. There was no apparent difference in TRAP+ cell numbers between PTH1R-deficient mutants and controls (Appendix Fig. 4B, C). Therefore, we believe that the decline in alveolar bone volume in PTH1R-ablated mice is mainly due to impaired bone apposition, not resorption. This imbalance of bone formation and resorption ultimately disrupted tooth eruption.
Impaired Periodontal Ligament (PDL) Differentiation in PTH1R-Deficient Mice
Normal development and maturation of PDL are indispensable for tooth eruption (Cho and Garant 2000). Because incisor PDL cells originate from progenitors that are Prx1+, we hypothesized that PTH1R deletion might affect PDL development. Indeed, Masson’s trichrome staining revealed that in Prx1Cre; PTH1Rfl/fl mutants, the PDL surrounding incisors was significantly narrowed. Furthermore, collagen fibers in mutant incisor PDL were irregularly aligned (Fig. 5A, B). Immunostaining with Periostin (POSTN), a marker for PDL fibers, indicated that mutants had significantly reduced POSTN expression in incisor PDL compared with controls (Fig. 5C). Despite no change in the radiolucent area surrounding incisors in µCT images (Fig. 2B, D), we observed sporadic bone-like tissue in mutant PDL adjacent to incisors. Almost two-thirds of mutants have bone-like tissue surrounding incisors at P7 and in all PDL of mutant incisors at P14 (Fig. 5D). The PDL of mutant molars remained unchanged (Fig. 5A, B).
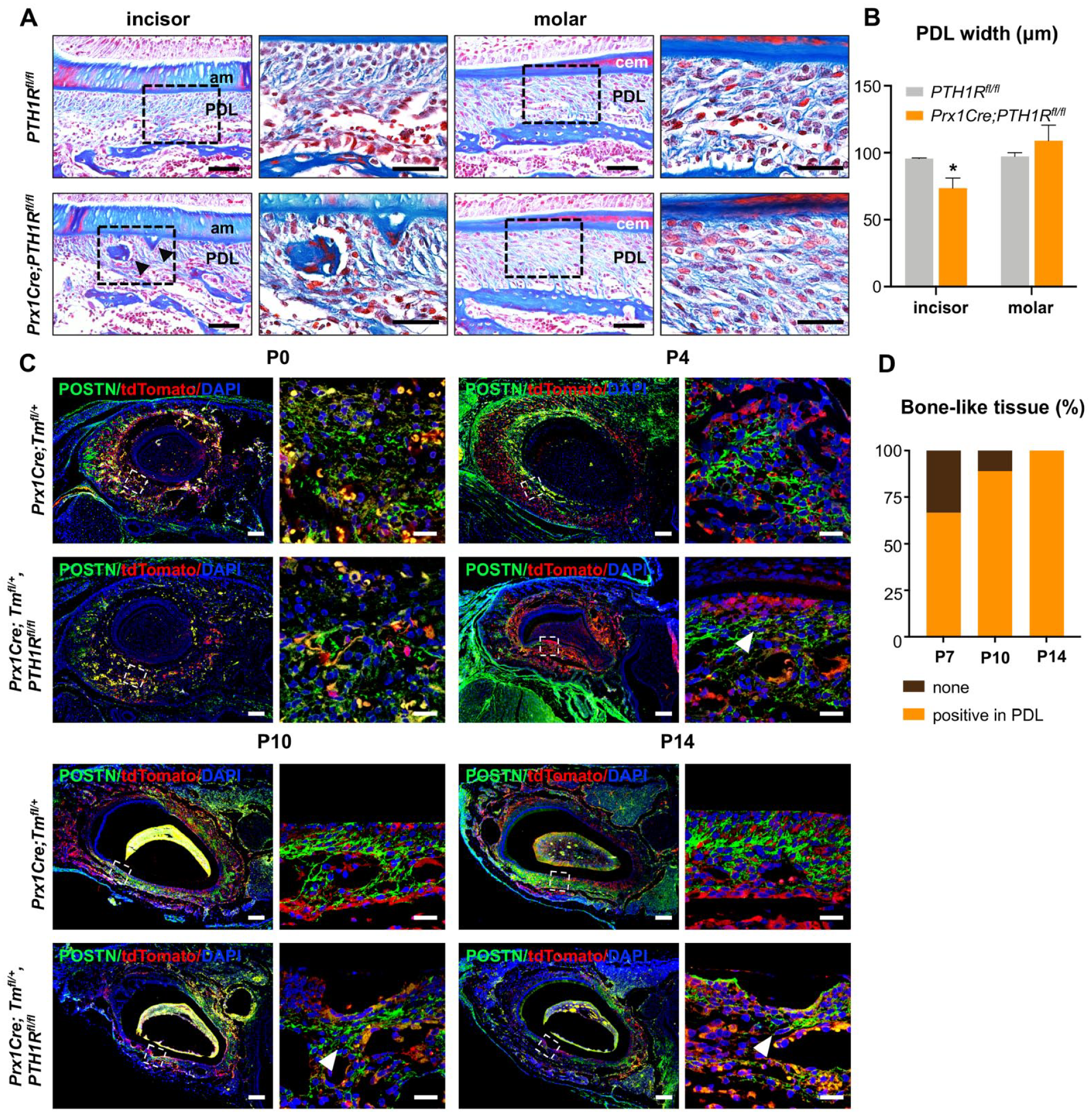
Impaired PDL differentiation in PTH1R-deficient mice. (
Discussion
Tooth eruption is a complex process involving many factors in which a tooth moves from the alveolar bone crypt into the oral cavity to a functional occlusal position (Wang 2013). The precise regulatory mechanism is widely debated and several theories have been suggested, including bone remodeling theory, dental follicle theory, root elongation theory, and hydrostatic theory (Wise and King 2008). Among these, molecular analysis of tooth eruption supports the bone remodeling theory as it suggests that a PTH1R gene mutation is associated with disturbances remodeling resulting in PFE (Frazier-Bowers and Hendricks 2015). An abundance of data demonstrate that alveolar bone growth provides the “motive force” to move the tooth throughout the eruption pathway. Significant alveolar bone growth occurs in the basal region of the alveolar bony crypt during eruption (Wise et al. 2007). Animal models with reduced alveolar bone development displayed eruption failure. MT1-MMP–/– mouse models and mice with siRNA targeted against Bmp6 both had impaired bone formation and failure in tooth eruption even though bone resorption was unaffected and an eruption pathway formed (Xu et al. 2016). We used a conditional deletion mouse model with PTH1R specifically ablated in Prx1+ cells located in a subset of craniofacial mesenchyme (Logan et al. 2002). They are predominately in mesenchyme tissue surrounding incisors and the base of molars and can differentiate into osteoblasts/osteocytes in alveolar bone, odontoblasts, and incisor PDL. PTH1R has been observed in dental mesenchymal cells, including DF cells, odontoblasts, and osteoblasts (Ono et al. 2016), overlapping mesenchyme-specific expression regions with Prx1+ cells. Ablating PTH1R caused a striking abnormality: the failure of mandibular incisor eruption. PTH1R-deficient mesenchyme progenitors at the base of molars also reduced eruption height in mutants, similar to clinical signs of PFE (Hanisch et al. 2018). Our data suggest that PTH1R signaling plays an essential role in alveolar bone development during eruption. First, we observed significantly reduced alveolar bone volume and bone growth rate concomitant with downregulated expression of key regulators for osteogenesis in PTH1R-ablated mice. Second, OMSCs residing in mandibular bone have differentiation abilities that directly affect alveolar bone remodeling (Matsubara et al. 2005; Akintoye et al. 2006). PTH1R-deleted OMSCs exhibited suppressed osteogenesis, suggesting that PTH1R expression in orofacial progenitor cells is indispensable for alveolar bone formation. Third, Prx1-expressing cells were not in molar PDL and root but rather in the alveolar bone at the base of molars. Delayed molar eruption suggests that reduced alveolar bone formation at the molar base affected eruption despite unaffected molar root and PDL development. Although previous work has focused largely on PTH1R’s function in root morphogenesis, our novel model sheds light on alveolar bone development and homeostasis during eruption.
It has been shown that formation of the eruption pathway relies on osteoclast recruitment and subsequent alveolar bone resorption (Wise and Fan 1989). Studies using murine osteopetrosis models (i.e., op/op mice, c-Fos and c-Src knockout mice) revealed eruption failure due to defects in osteoclast formation (Lowe et al. 1993; Grigoriadis et al. 1994). We did not observe differences in bone resorption between mutants and controls, consistent with studies using PTHrP-null mice or ablation of PTH1R in PTHrP+ cells (Philbrick et al. 1998; Takahashi et al. 2019). Also, this agrees with clinical observations in PFE where an eruption pathway was created but the tooth failed to reach the occlusal plane (Frazier-Bowers and Rhoads 2010). Therefore, we suggest that the reduced alveolar bone volume observed in mutants was mainly due to suppressed bone formation, leading to an imbalance in bone remodeling and subsequent eruption failure.
Prx1-expressing cells responsible for the differentiation of PDL cells are present in the DF region of mandibular incisors. Proper development and maintenance of fibrous PDL is critical in the supraosseous phase of tooth eruption (Cahill and Marks 1980). Many factors affect the mineralization state of entheses, and aberrant mineralization, such as that caused by elevated Wnt signaling, may lead to ankylosis (Wu et al. 2019). Mice lacking PTH1R in Osx+ cells showed no PDL formation, with the root cementum ankylosed to adjacent bone (Ono et al. 2016). PTH1R deficiency in PTHrP+ DF led to severely underdeveloped PDL (Takahashi et al. 2019). We observed thinner, less organized PDL with reduced POSTN expression in PTH1R-deleted cells, suggesting that PTH1R is required for differentiation of POSTN-expressing PDL cells. We observed sporadic mineralization nodes in mutant incisor PDL resembling those in mice with PTH1R-deficiency in PTHrP+ cells (Takahashi et al. 2019). This may be due to impaired cell differentiation or alveolar bone apposition, but the mechanism requires further investigation. We should mention that PFE differs from ankylosis. PFE does not exhibit cementum-bone fusion. Surgeons extracting teeth in PFE patients report tooth mobility in the socket (Proffit and Vig 1981). We found the radiolucent area in µCT unchanged and decreased alveolar bone volume surrounding incisors, indicating the tooth was not ankylosed. No phenotype was observed in enamel volume or amelogenesis related genes of Prx1Cre;PTH1Rfl/fl. Thus incisor eruption failure was mainly due to defects in alveolar bone and PDL development.
PTHrP exerts epithelial-mesenchymal paracrine/autocrine functions during tooth development. It is expressed in the enamel organ, whereas PTH1R is expressed in adjacent mesenchymal components and alveolar bone, suggesting paracrine action during eruption (Philbrick et al. 1998). Recently, researchers using PTHrP-CreER mice discovered PTHrP-expressing cells in molar DF, which express PTH1R. Conditional inactivation of PTH1R in PTHrP+ DF revealed an autocrine function of PTHrP-PTH1R signaling (Takahashi et al. 2019). However, Prx1+ cells were found primarily in the mesenchymal compartment of alveolar bone surrounding incisors and under molars, which are PTHrP-negative regions. Since PTH1R is expressed in Prx1+ mesenchymal cells, the paracrine function of PTHrP is nullified in these cells in Prx1Cre;PTH1Rfl/fl mice. It appears that the autocrine function of PTHrP-PTH1R in molar DF remained, as we did not observe alterations in molar root and PDL development as Takahashi et al. previously reported.
Our studies have some limitations. First, results showing arrested incisor eruption but incomplete impairment of molar eruption may not ideally reflect human PFE conditions that predominantly affect molars rather than anterior teeth (Frazier-Bowers et al. 2010). The eruption process continues throughout life in mouse incisors in contrast to tooth development and eruption in humans. Second, Prx1Cre is expressed in limb bud mesenchyme, so conditional deletion of PTH1R was not restricted to craniofacial regions. The axial skeleton of Prx1Cre;PTH1Rfl/fl mice is unaffected, and they survive relatively longer than Osx-PTH1R mutants. Approximately 80% of mutants survive weaning and 30% survive 6 mo. Previously, lack of good model systems limited investigations of tooth eruption. We believe our transgenic mice identify putative stem cells with eruption problems and may decipher genetic mechanisms of PTH1R signaling.
Our data reveal the important role of PTH1R signaling in orofacial mesenchymal stem cells for modulating alveolar bone development and maintaining proper periodontal ligament function during eruption. Using this conditional deletion mouse model to understand phenotypes caused by PTH1R mutations may provide new insights into its function in the pathogenesis of PFE.
Author Contributions
C. Cui, contributed to conception, design, data acquisition, and analysis, drafted and critically revised the manuscript; R. Bi, W. Liu, S. Guan, contributed to data acquisition and interpretation, critically revised the manuscript; P. Li, contributed to data acquisition and analysis, critically revised the manuscript; D. Song, R. Xu, L. Zheng, Q. Yuan, contributed to data analysis, critically revised the manuscript; X. Zhou, contributed to conception and design, drafted and critically revised the manuscript; Y. Fan, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
DS_10.1177_0022034520934732 – Supplemental material for Role of PTH1R Signaling in Prx1+ Mesenchymal Progenitors during Eruption
Supplemental material, DS_10.1177_0022034520934732 for Role of PTH1R Signaling in Prx1+ Mesenchymal Progenitors during Eruption by C. Cui, R. Bi, W. Liu, S. Guan, P. Li, D. Song, R. Xu, L. Zheng, Q. Yuan, X. Zhou and Y. Fan in Journal of Dental Research
Footnotes
Acknowledgements
We thank Dr. Beate Lanske and Dr. Henry M. Kronenbergb for providing PTH1Rfl/fl mice and the valuable help in this project. We thank Mr. Michael J. Densmore for assistance with editing the manuscript.
A supplemental appendix to this article is available online.
This study was supported by NSFC grants 81800928, Young Elite Scientist Sponsorship Program by CAST 2018Q NRC001, China Postdoctoral Science Foundation (2018T110982 and 2018M631090), Sichuan Science and Technology Program (No. 2019YJ0054) to Yi Fan.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.