
Abstract
The presence of periodontal diseases (PDs) often strongly correlates with other severe chronic inflammatory conditions, including cardiovascular disease, diabetes, and arthritis. However, the mechanisms through which these diseases interact are unclear. In PD, tissue and bone destruction in the mouth is driven by elevated recruitment of polymorphonuclear neutrophils (PMNs), which are primed and recruited from the circulation to sites of inflammation. We predicted that systemic effects on PMN mobilization or priming could account for the interaction between PD and other inflammatory conditions. We tested this using a mouse model of ligature-induced PD and found elevated PMN counts specifically in bone marrow, supporting a systemic effect of periodontal tissue inflammation on PMN production. In contrast, mice with induced peritonitis had elevated PMN counts in the blood, peritoneum, and colon. These elevated counts were further significantly increased when acute peritonitis was induced after ligature-induced PD in mice, revealing a synergistic effect of multiple inflammatory events on PMN levels. Flow cytometric analysis of CD marker expression revealed enhanced priming of PMNs from mice with both PD and peritonitis compared to mice with peritonitis alone. Thus, systemic factors associated with PD produce hyperinflammatory PMN responses during a secondary infection. To analyze this systemic effect in humans, we induced gingival inflammation in volunteers and also found significantly increased activation of blood PMNs in response to ex vivo stimulation, which reverted to normal following resolution of gingivitis. Together, these results demonstrate that periodontal tissue inflammation has systemic effects that predispose toward an exacerbated innate immune response. This indicates that peripheral PMNs can respond synergistically to simultaneous and remote inflammatory triggers and therefore contribute to the interaction between PD and other inflammatory conditions. This suggests larger implications of PD beyond oral health and reveals potential new approaches for treating systemic inflammatory diseases that interact with PD.
Introduction
Periodontal disease (PD) affects between 20% and 50% of the global population with growing evidence supporting its association with other inflammatory diseases, including heart disease, arthritis, and diabetes (Lakschevitz et al. 2011; Leech and Bartold 2015; Chang et al. 2020). Several studies have shown how untreated PD leads to increased medical care costs for nonoral conditions, including patient hospitalization rates (Jeffcoat et al. 2014). The interaction of inflammatory diseases with PD suggests a shared, underlying pathology that may be exploited to better manage patients and reduce the economic burden. Although the underlying biopathological mechanism is as yet unclear, we hypothesize that PD may prime the immune system, thereby influencing the response to a subsequent stimulus.
Polymorphonuclear neutrophils (PMNs) are first-line innate immune defenders during acute and chronic inflammation (Nauseef and Borregaard 2014) and are recruited constitutively to healthy mucosal sites, including the colon and oral cavity (Landzberg et al. 2015; Fine et al. 2016). During an inflammatory response, cytokines produced at the local site enter the bloodstream and upregulate PMN production in the bone marrow (BM), mobilization, and priming of circulating PMNs (Fine et al. 2019). These primed PMNs are now able to more efficiently enter tissues across the endothelial barriers both in healthy mucosal tissues and at sites of inflammation. While PMNs are essential for immune surveillance and protection, they can also cause damage to host tissues, as observed in PD (Weiss 1989; Sima et al. 2016).
Constitutive recruitment of PMNs to the healthy oral cavity in humans serves to contain the commensal biofilm at the interface between the gingival crevice and the tooth. In PD, oral PMN levels are greatly increased (Landzberg et al. 2015). Oral inflammation is associated with a hyperactive oral PMN phenotype (Aboodi et al. 2011; Hajishengallis et al. 2015; Fine et al. 2016) characterized by elevated phagocytosis and degranulation, as well as greater production of reactive oxygen species and neutrophil extracellular traps. This hyperactive PMN response to oral pathogens is accompanied by an upregulation of proinflammatory cytokines in tissues and the circulation (Figueredo et al. 2005). Hyperfunctionality of blood PMNs in PD patients compared to healthy or treated controls has been noted (Johnstone et al. 2007; Matthews et al. 2007), but any PMN-centric crosstalk between PD and other inflammatory conditions has not been tested. Conversely, excessive recruitment and “dysregulated” function of PMNs is known to be implicated in a wide range of chronic inflammatory diseases (Eksioglu-Demiralp et al. 2001; Karima et al. 2005; Garcia-Romo et al. 2011; Bruijnzeel et al. 2015; Sagiv et al. 2015).
Because PMN mobilization and priming occur throughout the body and not just at the inflamed tissue site, there is opportunity for misdirected or unwanted PMN-mediated collateral damage at unrelated secondary sites. It has previously been shown that mice with chronic inflammatory conditions, including colitis, diabetes, and lung inflammation, have increased PMN recruitment to an unrelated secondary peritoneal infection (Bian et al. 2012). However, the role of PMNs in the interaction between PD and a second inflammatory insult has not been tested. Thus, due to their important role as innate immune responders, their ubiquity/abundance, and their high sensitivity to diverse proinflammatory signals, we propose that PMNs might play a critical role as a potential nexus for crosstalk between inflammatory diseases that interact with PD.
We have developed a sensitive test for assessment of priming of PMNs based on upregulation of specific cell surface markers on peripheral PMNs in mice and human, which we used previously to track PMN priming responses in BM and blood during the course of self-resolving peritonitis (Fine et al. 2019). Here we use this flow cytometric testing approach to determine the effects of underlying PD on PMN responses over the time course of a secondary self-resolving peritoneal challenge.
Methods
Mouse Models
Mouse studies were performed in accordance with all relevant ethical regulations and were approved by the University of Toronto Animal Care Committee and the Research Ethics Board (Protocol #20010664). PD was induced by surgical installation of a nylon ligature as described previously (Chadwick and Glogauer 2020), and mice were used in experiments after 1 wk. Control mice without ligature-induced PD were sacrificed on day 7, along with PD mice. Peritonitis was induced by intraperitoneal injection of pHrodo-Red Escherichia coli BioParticles (Molecular Probes) as described previously (Fine et al. 2019). BM was flushed from 1 femur per mouse using 3 mL cold phosphate-buffered saline (PBS). Blood was recovered by cardiac puncture using EDTA (50 mM) as an anticoagulant and then diluted with 2 volumes of cold PBS. Cells were retrieved from the peritoneal cavity by lavage with 3 mL cold PBS. Colonic PMNs were obtained by gently scraping the luminal surface of the colon after it had been dissected. This was performed while the colonic tissue was immersed in cold PBS (3 mL). Oral PMNs were recovered by three 100-µL rinses with cold PBS as described previously (Chadwick and Glogauer 2020). Rinses containing colonic and oral PMNs were filtered through a 40-µm mesh filter. Cell counts were obtained on a Coulter counter, and the counts were normalized to the volume recovered. Cell counts for BM are reported “per femur.” Cell counts for blood are reported per 100 µL. Lavage fluid counts are per peritoneum, per colon, and per oral cavity, respectively. All mice were 8 to 16 wk old C57BL/6 and were euthanized by CO2 inhalation. Ex vivo BM experiments were performed as follows. Then, 250 µL flushed BM in PBS was aliquoted into 4-mL flow cytometry tubes and incubated for 30 min at 37°C. After ex vivo incubation, whole BM cells were fixed and labeled for flow cytometry. PMNs were gated based on expression of Ly6G.
Human Gingivitis Study
Healthy human volunteers were instructed to cease oral hygiene practices, including brushing and flossing, for 3 wk (gingivitis induction phase), followed by a 2-wk recovery period. Blood and saliva were collected during the nonbrushing regime (day 4, day 7, day 14, and day 21). Testing was also done after reinstitution of proper oral hygiene practices (day 28 and day 35). For blood stimulation assays, 100 µL fresh human blood was incubated for 30 min at 37°C, in the presence or absence of formyl-Met-Leu-Phe (fMLF; Sigma-Aldrich). Samples were fixed and labeled for flow cytometry. Blood plasma aliquots were frozen at −80°C for subsequent analysis of cytokines. For further details on human periodontal disease subjects and gingivitis subjects, see the Appendix.
Cytokine Analysis
Multiplex cytokine analysis was performed using High Sensitivity Cytokine Array kits (Randox Laboratories) and analyzed on the Evidence Investigator Biochip system (Randox Laboratories) according to the manufacturer’s instructions.
Statistical Analysis
All P values were determined by 1-way analysis of variance (ANOVA) with a post hoc Fisher’s least significant difference (LSD) test for multiple comparisons or 2-way Student’s t test unless indicated otherwise. P ≤ 0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism software (version 7.00; GraphPad Software).
Results
PMN Recruitment to the Oral Cavity
To determine whether PMNs play a role in the crosstalk between PD and an independent inflammatory condition, we used an established mouse model of ligature-induced periodontal disease (Chadwick and Glogauer 2020). This model allowed us to overlay an acute peritonitis in a system that is highly tractable, in which immune cells can be harvested from various tissues. PMNs are recruited constitutively to the healthy mucosal surface of the oral cavity, and this process is greatly accelerated in PD (Landzberg et al. 2015; Fine et al. 2016). We confirmed this by testing oral PMN counts from healthy human volunteers (n = 35) and from patients with stage 2 grade A/B PD (n = 37). We found that the mean number of PMNs recovered from the oral cavity in healthy volunteers was 2 × 106 PMNs, while 7 × 106 PMNs were harvested from patients with PD (Fig. 1A). Next, we tested PMN recruitment to the oral cavity in control mice and in the mouse model of ligature-induced periodontal disease (Sima et al. 2016). We found that the mean count of PMNs recovered from the oral cavity in control mice was 2.5 × 104 PMNs, while 1.5 × 105 PMNs were recovered from the oral cavities in PD mice (Fig. 1B, C). Thus, ligature-induced PD in mice mimics human PD at the level of oral PMN recruitment.
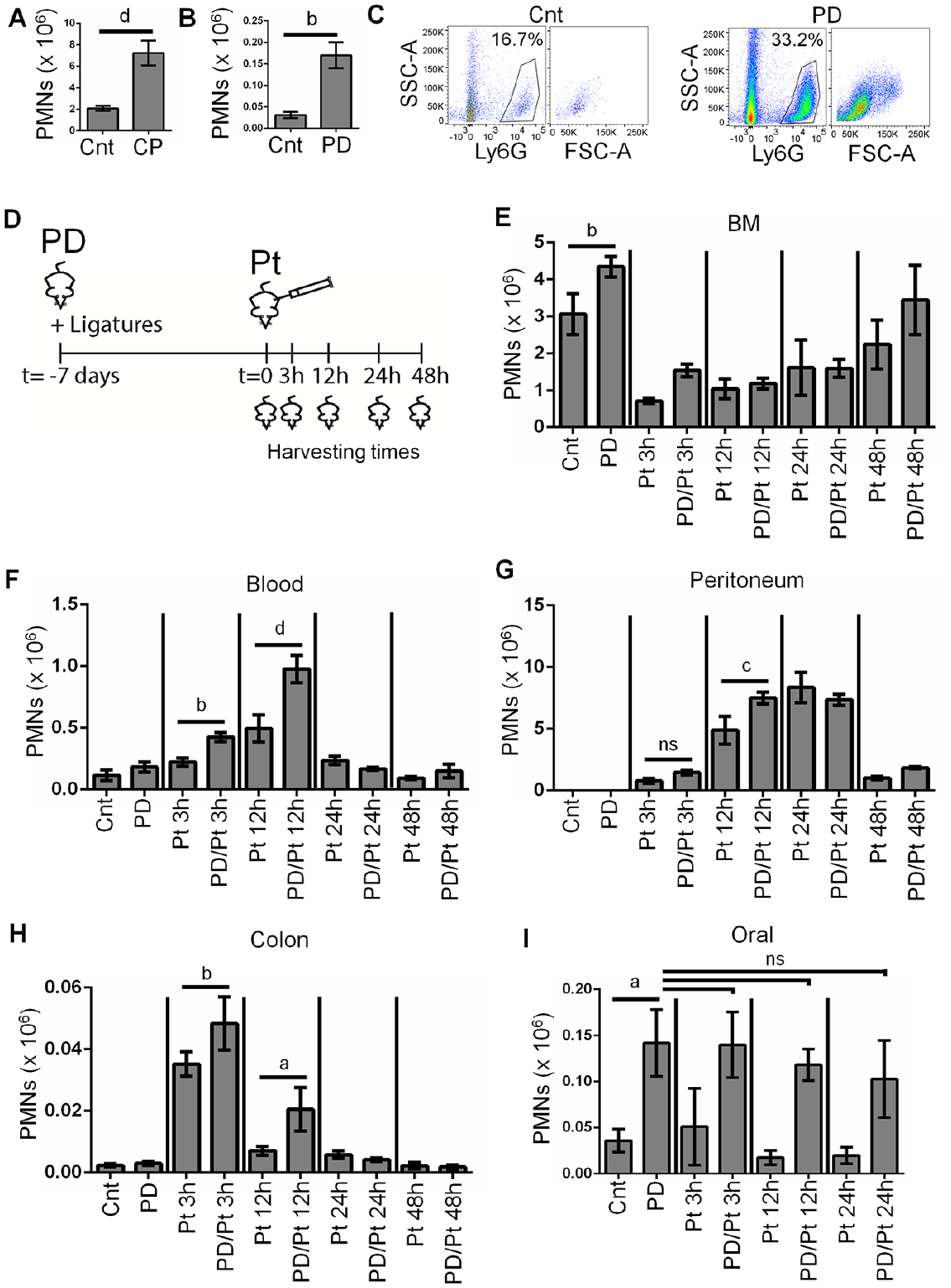
Periodontal disease (PD) enhances the polymorphonuclear neutrophil (PMN) response to a severe acute peritonitis. (
PD Enhances the PMN Response to Acute Peritonitis
Because PD is associated with other inflammatory diseases, we hypothesized that systemic PMN counts and/or activation state induced by PD could affect the innate immune response to an unrelated secondary inflammatory insult. To test this, we developed a tunable mouse model of inflammation by intraperitoneal injection of increasing doses of pHrodo-labeled E. coli particles (Fine et al. 2017), ranging from 2 to 100 µL (Appendix Fig. 1). We noted a continuous increase in PMN-associated responses (i.e., PMN counts, expression of a panel of CD markers of PMN activation states, and phagocytosis by peritoneal PMNs), which was directly proportional to the dose of peritonitis (Appendix Fig. 1). Based on this, we selected a low (2 µL) and a high (100 µL) dose for further studies.
We tested the effect of high-dose pHrodo (100 µL) on PMN levels in the BM, blood, peritoneum, oral cavity, and colon (a healthy mucosal site with known PMN accumulation) in the presence or absence of PD (Fig. 1D). In the absence of peritonitis, ligature-induced PD produced increased PMN counts in the oral cavity and in BM (Fig. 1E, I). This indicates both local and systemic effects of oral inflammation on the innate immune system. In contrast, there was no effect on steady-state PMN counts in blood, peritoneum, or colon in PD mice compared to controls (Fig. 1F–H). To study the potential interaction between this PD-induced innate immune priming and a secondary infection, we tested a time course spanning the acute and resolution phases of peritonitis. Upon induction of peritonitis alone (Pt), PMN counts in BM were significantly reduced within 3 h compared to control mice, consistent with a concomitant increase in PMN counts in the blood, peritoneum, and colon. However, when peritonitis was induced in mice that also had PD (PD/Pt), there were significantly greater levels of PMNs in the blood (3 and 12 h), peritoneum (12 h), and colon (3 and 12 h) compared to mice that had peritonitis only.
To determine if the severity of the acute inflammation accounted for the synergistic activation of the innate immune system at 3 and 12 h, we tested whether a mild peritonitis (2 µL of bacteria into peritoneum; Fig. 2) would also induce a similar increase in PMN recruitment in conjunction with PD. We found that reduction of PMNs in the BM with mild peritonitis was not as pronounced as it was with the severe peritonitis (Fig. 2A). PMN counts in the peritoneum were elevated at 3 h and continued to increase at 12 h, while PMNs in blood peaked at 3 h and had resolved by 12 h (Fig. 2B, C), consistent with more rapid resolution of peritonitis in response to the low dose of pHrodo. However, as with severe peritonitis, low-dose peritonitis in mice that had underlying PD resulted in elevated PMN counts in the blood (3 h) and peritoneum (12 h), as compared to mice that did not have PD. Thus, the underlying presence of PD has a priming effect on the innate immune response to peritonitis, regardless of severity, in mice.
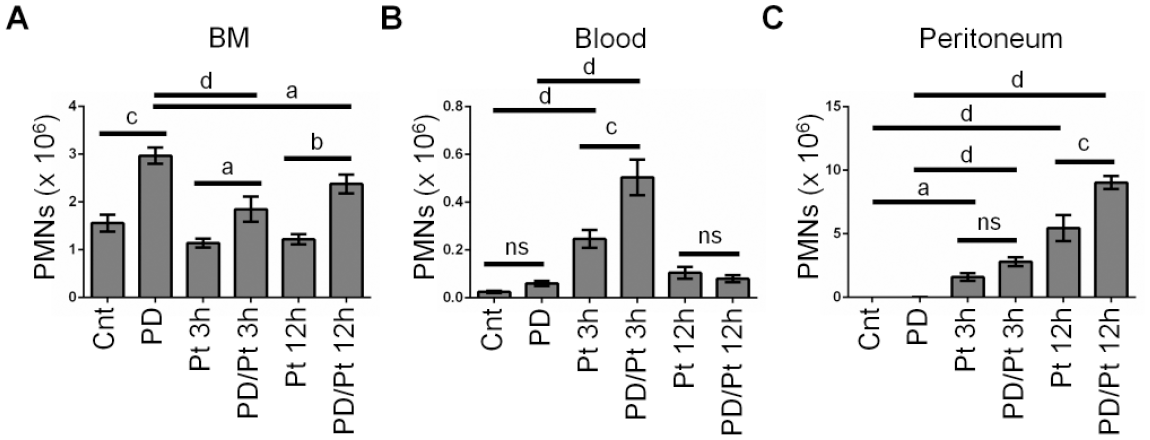
Periodontal disease (PD) enhances the polymorphonuclear neutrophil (PMN) response to a mild acute peritonitis. Healthy control (Cnt) mice, PD mice, low-dose peritonitis (Pt) mice, or double-hit (PD/Pt) mice with PD and acute low-dose peritonitis were sacrificed at the indicated time points. A total of 2 µL Escherichia coli–pHrodo was used to induce acute peritonitis. Tissue compartments were acquired and assessed by flow cytometry. The percentage of Ly6G+ve/F480−ve cells in each compartment, combined with total cell counts, was used to determine the absolute PMN count in bone marrow (
PD Does Not Alter Macrophage Responses to Peritonitis
To determine whether the effect was PMN specific, we analyzed macrophage levels, which have been proposed to play a role in the response to inflammation at distant sites (Hoyer et al. 2019). Control and PD mice had similar F4/80+ve macrophage counts in the peritoneal cavity (Appendix Fig. 2A, B). At each time point, there was no difference between macrophage counts or phagocytosis of pHrodo particles between Pt and PD/Pt mice (Appendix Fig. 2C). These results suggest that the presence of PD does not alter peritoneal macrophage levels in response to peritonitis.
PD Enhances Ex Vivo Changes in PMN CD Marker Expression
We have shown that combining PD with a secondary infection enhances tissue recruitment of PMNs. To determine whether the activation state of the PMNs is altered, we analyzed steady-state CD marker expression on PMNs in BM, blood, and peritoneum. We performed multicolor flow cytometry with gating on PMNs to assess the surface expression of CD11b (αM-integrin), CD66a (CEACAM1), and CD62L (L-selectin), markers that we and others have shown to be associated with PMN activation (Zhang et al. 2015; Fine et al. 2019). CD66a and CD11b are contained in PMN granule membranes, and upregulated surface expression indicates degranulation, while CD62L is shed with aging. All 3 of these surface markers are involved in PMN adhesion and tissue recruitment (Skubitz et al. 1996; Green et al. 2004). We did not see a difference in PMN expression of these 3 CD markers in control mice compared to PD mice in BM, blood, or peritoneum (Fig. 3 and Appendix Fig. 3). Furthermore, the levels of these markers were similar at each time point and in each tissue, whether mice were exposed to peritonitis in the presence or absence of PD. Therefore, we hypothesized that the PMNs may be primed to become activated and we could assess this ex vivo. Whole BM from control mice, PD, Pt (12 h), and PD/Pt (12 h) mice was incubated at 37°C for 30 min to induce activation or maintained on ice. We found that incubating BM PMNs from PD/Pt mice at 37°C resulted in significantly greater upregulation of CD66a and CD11b, as well as a greater downregulation of CD62L, than BM from control mice or single PD or peritonitis mice (Fig. 3J–L). This indicates that PMNs from PD/Pt mice are primed for greater ex vivo activation compared to the other 3 groups.
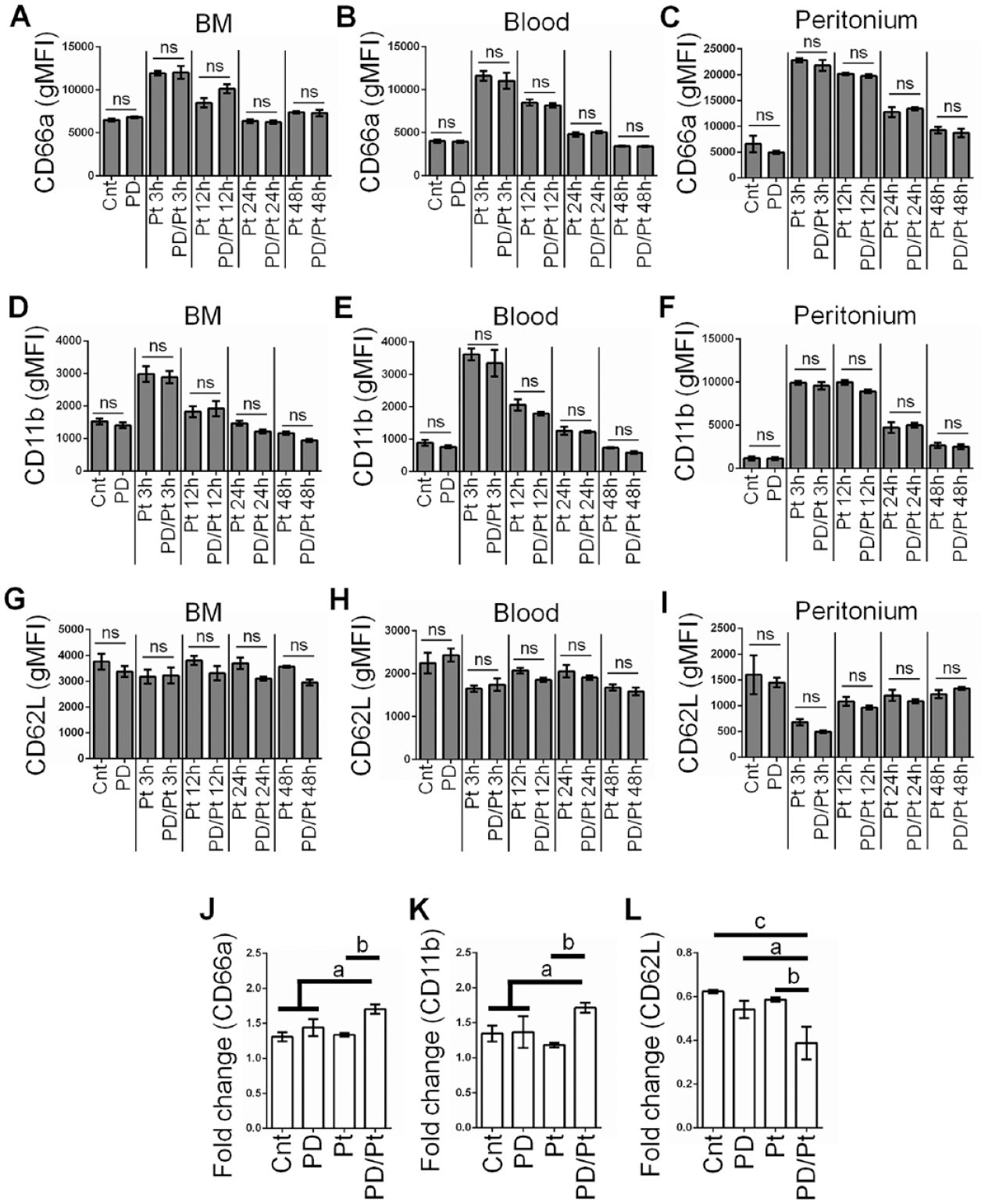
Periodontal disease (PD) combined with peritonitis enhances ex vivo polymorphonuclear neutrophil (PMN) responses. (
Human Gingival Inflammation Enhances PMN Ex Vivo Activation
To test whether PD also affects circulating PMNs in humans, we used an experimental gingivitis model and tested blood PMN activation through upregulation of CD11b. Subjects suspended normal oral hygiene (OH) practices for a period of 3 wk, followed by resumption of OH. During the gingivitis regime, clinical parameters including bleeding on probing (BOP), gingival index (GI), and oral PMN load were significantly increased and largely returned to baseline levels with resumption of OH (Fig. 4A–C). Using fresh blood samples acquired from gingivitis subjects at each visit, we analyzed CD11b expression in response to ex vivo stimulation by 10 µM fMLF, a bacterial peptide that is a strong stimulus for PMNs. We found an elevated fold-increase in CD11b expression during the gingivitis regime, which reverted to baseline after 2 wk of resumption of oral hygiene practices (Fig. 4D, E). Thus, human induced-gingivitis enhances priming of PMNs in circulation.
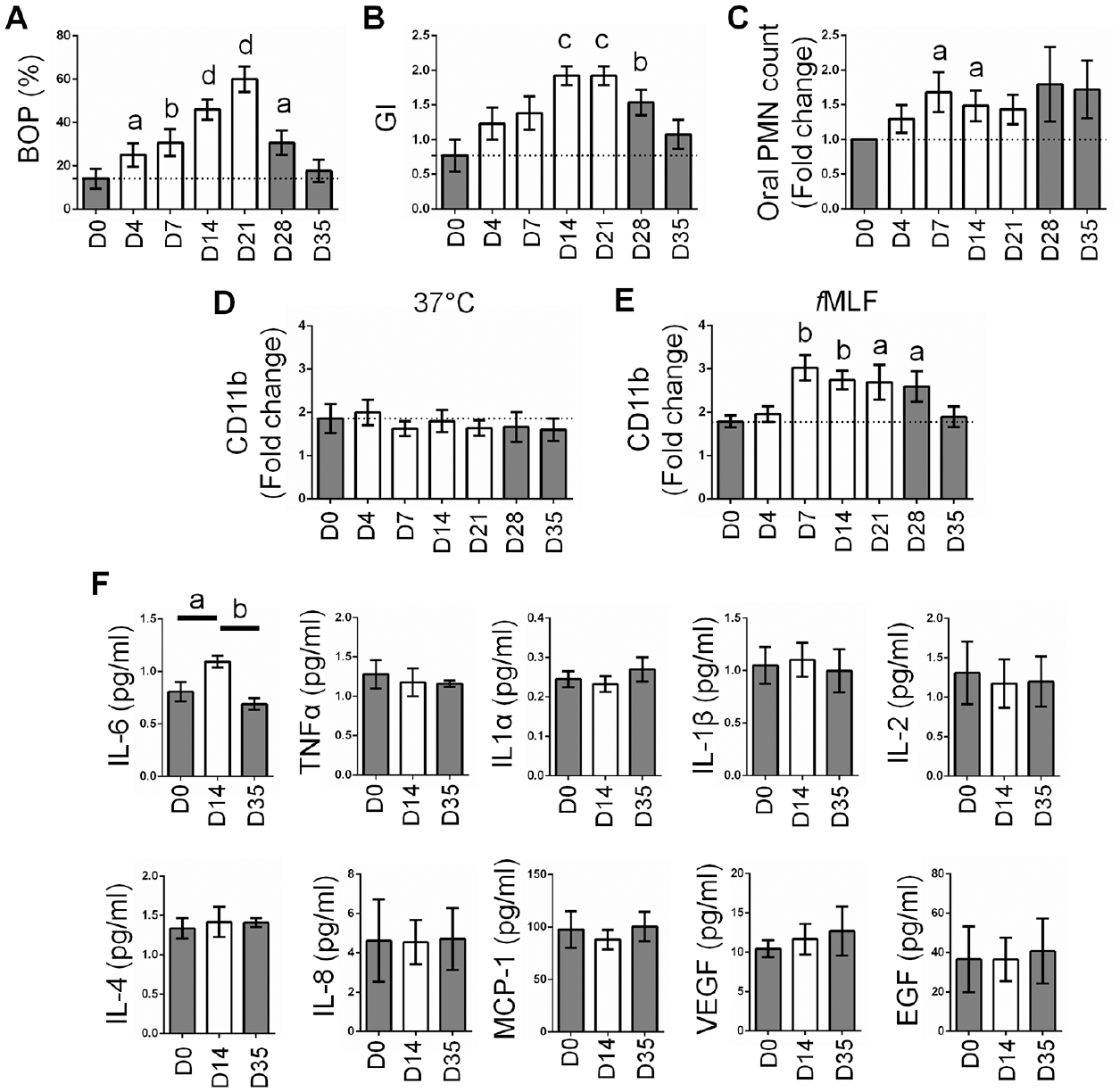
Gingival inflammation enhances ex vivo responses of blood polymorphonuclear neutrophils (PMNs). Healthy volunteers were recruited (n = 13) and ceased oral hygiene for a period of 21 d. Blood and saliva were collected on days 0, 4, 7, 14, and 21 of the nonbrushing regime and days 28 and 35 after reinstating oral hygiene practices. (
Based on increased ex vivo priming of blood PMNs from gingivitis subjects, we hypothesized that proinflammatory factors in the blood could be responsible for this effect. We therefore tested a standard array of proinflammatory cytokines in blood plasma of these subjects, using multiplex sandwich enzyme-linked immunosorbent assay (ELISA). Cytokines were tested prior to cessation of hygiene (day 0), on week 2 of the gingivitis regime, and on week 2 after resumption of oral hygiene practices. Only interleukin (IL)–6, a granulopoietic factor that plays a critical role in the acute inflammatory response, showed a significant increase in plasma of patients during gingivitis, which reverted to normal levels after reinstitution of brushing (Fig. 4F).
Discussion
The mechanisms underlying “crosstalk” between PD and other inflammatory diseases are not known. We present evidence in both mouse and human PD models showing that oral inflammatory disease can alter PMN counts and priming potential systemically, so that a hyperinflammatory PMN response occurs in the presence of an unrelated second inflammatory trigger. Hence, the synergistic interaction of 2 simultaneous inflammatory diseases could be a result of convergence of signaling to promote PMN-mediated hyperinflammatory innate immune responses (Fig. 5).
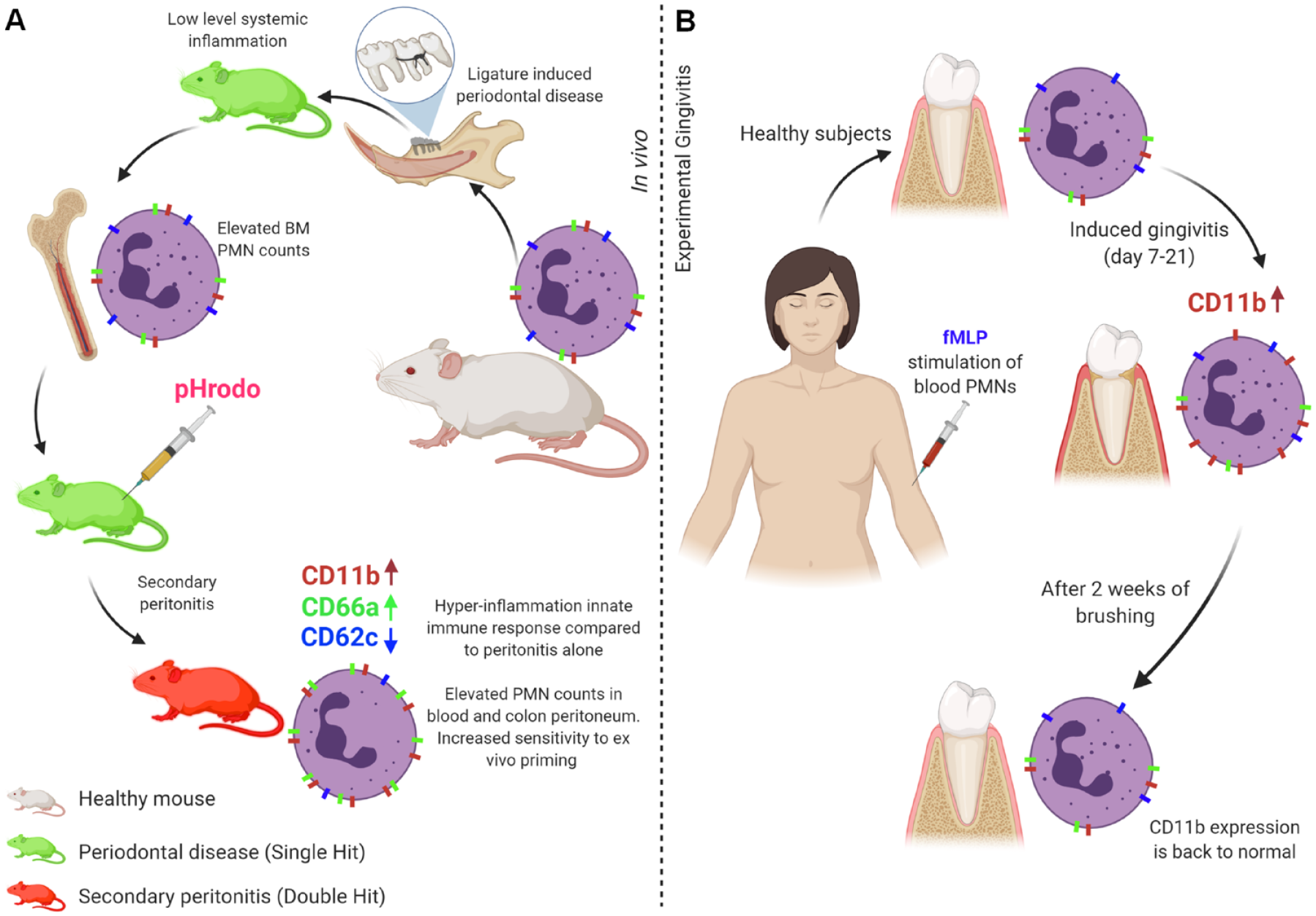
Periodontal disease (PD) primes the innate immune system. PD produces systemic effects on polymorphonuclear neutrophils (PMNs) in mice (
In addition to increased oral PMN counts, we found that mice with ligature-induced PD had increased PMN counts in the BM but not in the blood, suggesting that baseline production of PMNs is increased due to signals from the chronically inflamed periodontium. In the mouse model of PD, the inflammatory insult has been in place for upward of 1 wk prior to PMN analysis, and the steady-state effects of this chronic inflammation are different from the short-term inflammatory burst associated with acute peritonitis. In our human gingivitis subjects, we have identified IL-6 as a factor that could be associated with the systemic effects of PD. IL-6 is a granulopoietic factor (Suwa et al. 2000) and an effective PMN priming agent (Biffl et al. 1994), and it is known to be associated with periodontal disease (McGee et al. 1998). Furthermore, serum IL-6 levels are higher in transplant patients with periodontitis, compared to those without (Machado et al. 2020). Further studies are necessary to identify granulopoietic and other factors that might also be involved in systemic effects of PD on PMN counts in the BM. Some potential factors include IL-23, IL-17, and granulocyte colony-stimulating factor (G-CSF), which are critical for granulopoiesis and PMN mobilization (Christopher and Link 2007), and they are upregulated in periodontal disease (Schenkein et al. 2010; Qi et al. 2013; Zhang et al. 2020). Hemorrhagic shock was shown to induce priming of BM PMNs toward a second inflammatory hit, and this occurred through an IL-23/IL-17/G-CSF signaling axis (Liu et al. 2009). Chemokines that regulate PMN retention (CXCL12/SDF-1) and egress (CXCL1/KC, CXCL2/MIP-2) from the BM are also potential factors of interest (Sadik et al. 2011). Other proinflammatory factors, including IL-1β, tumor necrosis factor α, IL-2, and interferon γ, have been shown to be elevated in serum samples of periodontal disease patients (Gorska et al. 2003), but these were not found to be elevated in our gingivitis subjects.
Our results suggest that, upon induction of acute peritonitis, PMNs are rapidly depleted from BM, resulting in a corresponding increase of blood and tissue PMNs. Concomitant with elevated PMN counts in the BM of PD mice relative to controls at baseline, PD mice showed a corresponding higher increase of blood and tissue PMNs compared to non-PD mice after peritonitis was induced. Interestingly, PMN recruitment to the colon, an unrelated mucosal site, was also increased. Although the colon is a tissue site in close proximity to the peritoneal cavity, elevated PMN counts in the colon closely mirrored increased PMN counts in the blood (3 and 12 h) but not increased counts in the peritoneum (3, 12, and 24 h), suggesting that this effect is not due to leakage from the peritoneum. These results suggest that increased PMNs in BM of PD mice can become mobilized and potentially respond to any inflammatory trigger at another site in the body, resulting in a synergistic hyperinflammatory response. Although our ex vivo studies indicate that crosstalk between 2 simultaneous inflammatory conditions (PD and Pt) enhances the capacity for subsequent activation, further studies are necessary to determine any differences in functionality of PMNs from the single- and double-hit models. One limitation of our study is that the mouse model of ligature-induced PD is not directly analogous to human disease, and therefore it would be of interest to confirm our results using other models, including a mouse model of periopathogen-induced PD and studies in nonhuman primates.
PMNs in the saliva, which emerge through the gingival sulcus, are expected to have an activated phenotype, relative to gingival tissue PMNs, due to interaction with the oral biofilm (Fine et al. 2016). We have been the first group to assess PMN load in the mouse oral cavity, and our approach will facilitate future studies to compare oral PMN phenotypes and responses in the gingiva and saliva using mouse models of oral disease. Our data support the concept that PMN responses are a nexus between concurrent inflammatory conditions. We have used mouse and human models to demonstrate that elevated PMN counts and activation/priming states occur as a direct result of overlapping inflammatory conditions. Therefore, priming of the immune system as a result of periodontal inflammation can influence the intensity of the innate immune response experienced at a second inflammatory site. Further studies are necessary to demonstrate whether this increased PMN-mediated inflammation contributes to increased specific clinical disease manifestations and mortality associated with comorbidities. Our study reinforces the ramifications of oral hygiene and oral health for systemic health and specifically implicates PMNs as an important axis for crosstalk between oral disease and other systemic inflammatory conditions. Health management organizations should therefore view prevention and treatment of PD as an essential outcome. Our results reinforce the proposal that host modulation therapy (Novak and Donley 2002; Steinberg et al. 2005), through suppression of PMN responses or PMN-secreted factors, is a potential therapeutic avenue in systemic inflammatory diseases that interact with PD.
Author Contributions
N. Fine, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; J.W. Chadwick, contributed to conception, design, data acquisition, and analysis, critically revised the manuscript; C. Sun, N. Khoury, contributed to data acquisition, critically revised the manuscript; K.K. Parbhakar, contributed to data acquisition and analysis, critically revised the manuscript; A. Barbour, M. Goldberg, H.C. Tenenbaum, contributed to data interpretation, drafted and critically revised the manuscript; M. Glogauer, contributed to conception, design, and data interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
DS_10.1177_0022034520963710 – Supplemental material for Periodontal Inflammation Primes the Systemic Innate Immune Response
Supplemental material, DS_10.1177_0022034520963710 for Periodontal Inflammation Primes the Systemic Innate Immune Response by N. Fine, J.W. Chadwick, C. Sun, K.K. Parbhakar, N. Khoury, A. Barbour, M. Goldberg, H.C. Tenenbaum and M. Glogauer in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by a Bone Team grant from the Canadian Institutes of Health Research to M. Glogauer (TBO-122068).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.