
Abstract
Despite the growing recognition of a host genetic effect on shaping gut microbiota composition, the genetic determinants of oral microbiota remain largely unexplored, especially in the context of oral diseases. Here, we performed a microbiome genome-wide association study in 2 independent cohorts of patients with oral squamous cell carcinoma (OSCC, n = 144 and 67) and an additional group of noncancer individuals (n = 104). Besides oral bacterial dysbiosis and signatures observed in OSCC, associations of 3 loci with the abundance of genus-level taxa and 4 loci with β diversity measures were detected (q < 0.05) at the discovery stage. The most significant hit (rs10906082 with the genus Lachnoanaerobaculum, P = 3.55 × 10–9 at discovery stage) was replicated in a second OSCC cohort. Moreover, the other 2 taxonomical associations, rs10973953 with the genus Kingella (P = 1.38 × 10–9) and rs4721629 with the genus Parvimonas (P = 3.53 × 10–8), were suggestive in the meta-analysis combining 2 OSCC cohorts. Further pathway analysis revealed that these loci were enriched for genes in regulation of oncogenic and angiogenic responses, implicating a genetic anchor to the oral microbiome in estimation of casual relationships with OSCC. Our findings delineate the role of host genotypes in influencing the structure of oral microbial communities.
Keywords
Introduction
Oral squamous cell carcinoma (OSCC), accounting for nearly 90% of oral cancer (Jemal et al. 2008), is a commonly occurring malignancy in the oral cavity attributed by genetic and environmental factors (Scully and Bagan 2009). These etiological parameters reveal that OSCC is of a multifactorial nature that, to some degree, alters or is affected by a specific microbial milieu in the oral cavity. Currently, this notion has been supported by extensive analyses of shifts in the composition and functionality of oral microbiome (Lim et al. 2017), implicating a role for oral microbes in OSCC through direct metabolism of carcinogens and inflammatory effects.
The community structure of human microbiota is affected by multiple factors, but the relative contribution of host genetics remains largely elusive. Recently, numerous microbiome genome-wide association studies (mGWAS) have suggested that host genotypes may play an important role in remodeling human microbiome composition (Blekhman et al. 2015; Bonder et al. 2016; Goodrich et al. 2016; Turpin et al. 2016; Wang et al. 2016; Igartua et al. 2017; Rothschild et al. 2018; Scepanovic et al. 2019; Hughes et al. 2020; Ishida et al. 2020; Kurilshikov et al. 2021), with most heritable taxa identified being gut commensals. Notably, it is found that microbial associations with host genetic variants are body site specific (Blekhman et al. 2015; Kolde et al. 2018). Although knowledge of the oral microbiome lags behind that of the gut, intriguing features of the microbial communities in the oral cavity are beginning to emerge. Just as in the gut, the community structure of the human oral microbiome is influenced by host genetic backgrounds (Blekhman et al. 2015; Kolde et al. 2018). Yet, the findings from twin studies aiming to scrutinize the heritable proportion of the oral microbiome in caries are somewhat paradoxical. Cariogenic taxa in saliva (Corby et al. 2005) and plaque (Corby et al. 2007) have been shown as inherited traits; however, other investigations found no association of potentially heritable bacteria with dental caries in children (Gomez et al. 2017) and adults (Papapostolou et al. 2011). These findings suggest that these heritable bacterial traits not only are relevant to the habitats of human microbiota but also connected with disease status. Given that in OSCC patients, a specific reservoir of oral microbiota resides in a unique ecosystem mainly attributed by environmental parameters and host inherited factors, determining the nature of host genotype–microbe interactions in oral cancer may offer a novel avenue to delineate the mechanisms of oral tumorigenesis.
We have previously demonstrated a relationship between oral microbiome and somatic mutational signatures in the tumors of oral cavity (Yang et al. 2018). Here, we interrogated the extent to which oral bacterial traits (diversity and genus-level abundance) are genetically driven in 2 independent cohorts of OSCC patients and an additional group of noncancer individuals by mapping microbiome quantitative trait loci (mQTL). Overall, our data reveal that oral dysbiosis is influenced by host genotypes at many loci encoding host carcinogenic and angiogenic pathway genes in OSCC.
Methods
Subject Recruitment and Sample Collection
Nonstimulated saliva and whole blood of 220 noncancer controls and 208 patients were collected for the discovery cohort in this study, with the approval by the institutional review board of Chung Shan Medical University Hospital, Taichung, Taiwan (CS1-20091). Cohort selection criteria and sample collection are described in detail in the Appendix. The replication cohort comprised 67 patients whose microbiota composition was reported previously (Lee et al. 2017) and analyzed in a second tier. This study was conducted in compliance with appropriate guidelines, with a Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist provided.
16S Ribosomal RNA Gene Sequencing and Data Processing
The detailed protocols for 16S ribosomal RNA (rRNA) gene sequencing and processing of sequence reads are described in the Appendix. In brief, the variable region 3–4 (V3–V4) of the small subunit rRNA (16S rRNA) gene was polymerase chain reaction (PCR) amplified and sequenced. Paired-end reads were processed by performing quality control checks, operational taxonomic units (OTU) clustering, taxonomy classification, and estimation of α and β diversity.
Genotyping
The genome-wide genotyping experiments were conducted by using the C2-58 Axiom Genome-Wide TWB 2.0 Array Plate (Thermo Fisher Scientific), and allelic discriminations of 4 selected single-nucleotide polymorphisms (SNPs) for replication were assessed by using the TaqMan assay with an ABI StepOne Real-Time PCR System (Applied Biosystems). Genotyping quality control (QC), variant calling, and filtering are described in detail in the Appendix.
mGWAS
At the discovery stage, mGWAS against 3 types of microbial traits, including α diversity, β diversity, and relative abundance of genus-level taxonomies, by using a linear mixed model in a cohort of 144 OSCC cases adjusted for sex and age was performed and is described in detail in the Appendix. In addition to a nominal genome-wide significance threshold (P < 5 × 10–8), the study-wide P value threshold was defined as a Bonferroni correction assuming 165,744 independent genetic association tests across 58 mGWASs (0.05 / (165,744 × 58) = 5.2 × 10−9). For the top 10 hits at the discovery stage, a verification analysis followed by a meta-analysis using a cohort of 104 noncancer controls with genome-wide genotyping data was performed. Associations in the same allelic direction with P < 0.05 in the replication or P < 5 × 10–8 in the meta-analysis were considered significant. In addition, to validate the host genotype–oral microbiome interactions exclusively for oral cancer, we performed a targeted replication analysis on top 4 hits of taxonomic associations followed by a meta-analysis using a previously reported cohort of 67 OSCC cases (Lee et al. 2017). In this disease-specific, targeted analysis, a significance P value threshold at 0.05 and 2.15 × 10–4 (0.05 / (4 × 58)) was used in the replication and meta-analysis, respectively.
Ingenuity Pathway Analysis
Pathway analysis of mQTLs was performed by using the Ingenuity Pathway Analysis (IPA; QIAGEN) and is described in detail in the Appendix.
Statistical Analysis
In addition to multiple significance thresholds applied in each stage, associations from mGWAS were adjusted by Benjamini–Hochberg correction. Unless otherwise stated, a P value of <0.05 was considered significant.
Results
Data Analysis Workflow and Association Summary
In this study, we collected salivary samples from a cross-sectional cohort of 428 unrelated subjects (Appendix Table 1), comprising 208 cases and 220 noncancer controls for 16S rRNA gene sequencing. Among them, host genotype data of 144 cases and 104 controls were obtained. To identify host genetic loci that influence the oral microbiome in OSCC, we conducted a genome-wide association analysis in 144 cases as the discovery stage (Fig. 1). Reproducibility of results was examined in 2 branches and then followed by meta-analysis: one implicating overall host genotype–microbe interactions in the oral cavity using a group of 104 noncancer controls and the other branch revealing a disease-specific link between host genetics and oral dysbiosis in OSCC using an additional cohort of 67 patients with oral cancer from a previous report (Lee et al. 2017).
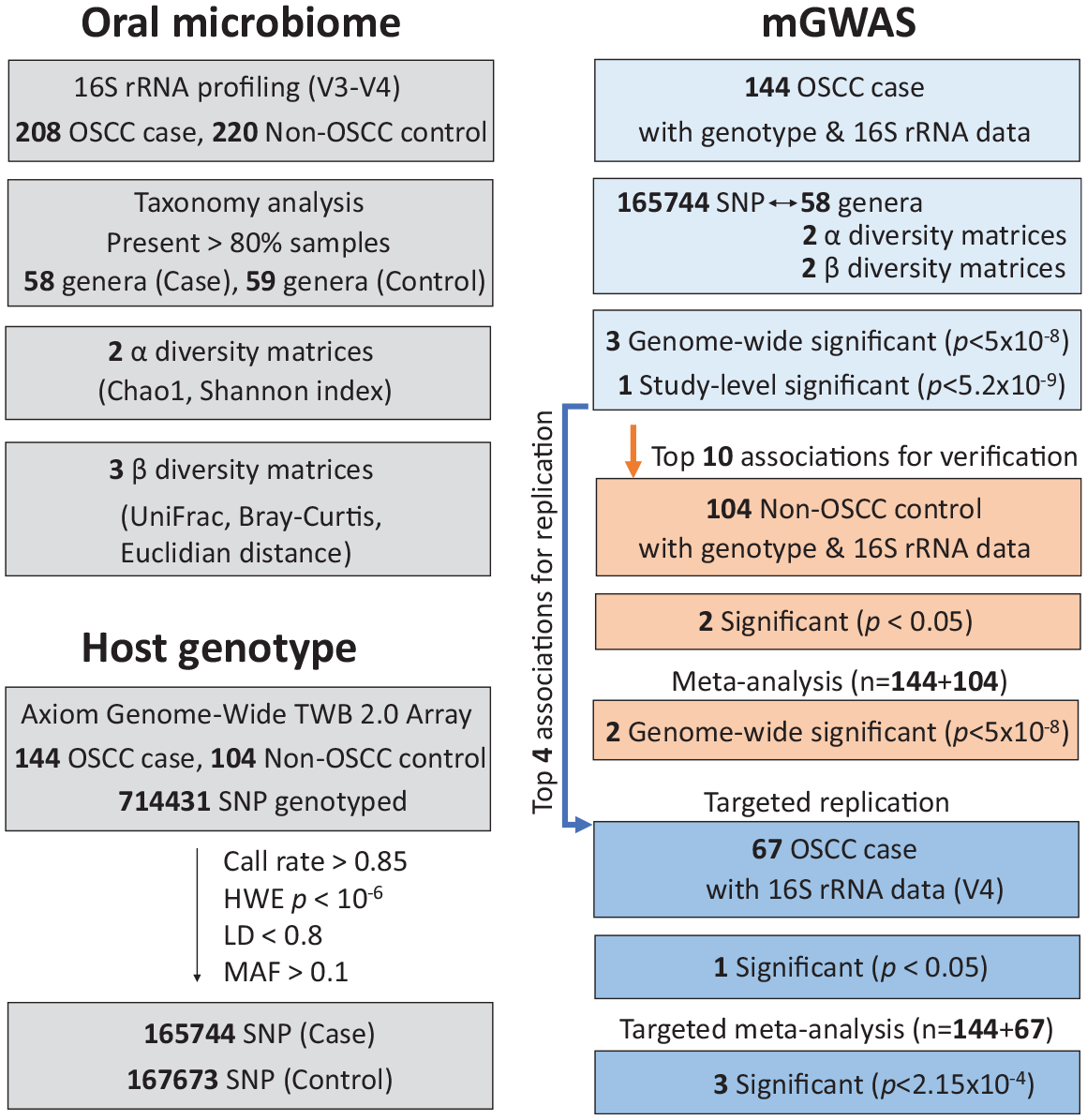
Flowcharts of study design and association summary. Analysis overview highlighting the data collected, steps taken, and selections made. mGWAS, microbiome genome-wide association study; OSCC, oral squamous cell carcinoma; SNP, single-nucleotide polymorphism.
Genetic Association of Oral Dysbiosis in OSCC
To detect association signals between host genetic variations and the oral microbiome in OSCC, we performed a genome-wide association analysis between 165,744 common SNPs and multiple microbial traits, including 58 genus-level taxonomies, 2 α diversity matrices, and 2 β diversity matrices using a cohort of 144 OSCC cases adjusted for sex and age as the discovery stage. In search of an association with bacterial taxa, our analysis identified 3 mQTLs at q < 0.05, all of which attained the genome-wide significance threshold (P < 5 × 10–8) (Fig. 2A). We did not detect a host genetic effect (q < 0.05) on bacterial α diversity measures (Appendix Table 2). In addition, 4 SNPs, albeit not at the genome-wide significance level, were found to be associated with β diversity (q < 0.05; Fig. 2B and Appendix Table 2).
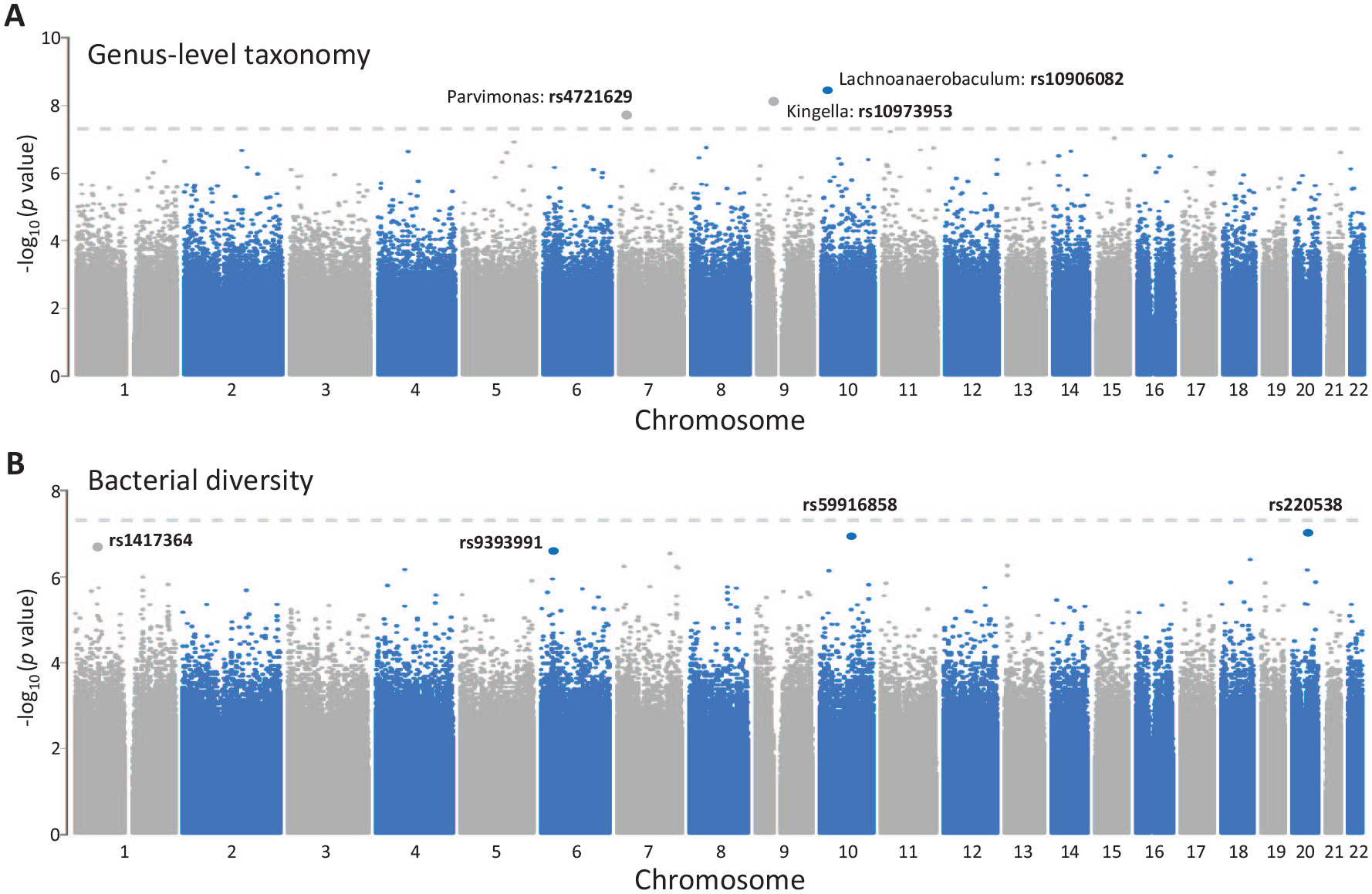
Genome-wide associations between host genotypes and oral microbiome in oral squamous cell carcinoma. The Manhattan plots presented for variants pruned for linkage disequilibrium (LD) (r2 < 0.8) show microbiome quantitative trait loci for the abundance of bacterial genera (
We then used 2 cohorts to validate top taxonomic associations detected from the discovery screening. Using a cohort of 104 noncancer subjects, despite the difference in malignant disease pathology relative to the discovery cohort, verification analyses confirmed 2 associations out of top 10 hits with the same allelic direction (Table): rs75808570 with the abundance of Ruminococcaceae UCG-014 and rs10496915 with the abundance of Dialister (P < 0.05 at verification and P < 5 × 10–8 in the meta-analysis as adjusted for sex, age, and disease status). These signals may represent universal host genotype–microbe interactions in oral cavity regardless of the disease state.
Microbiome Quantitative Trait Loci Mapping Results of Oral Microbiome in OSCC Cases and Noncancer Controls.
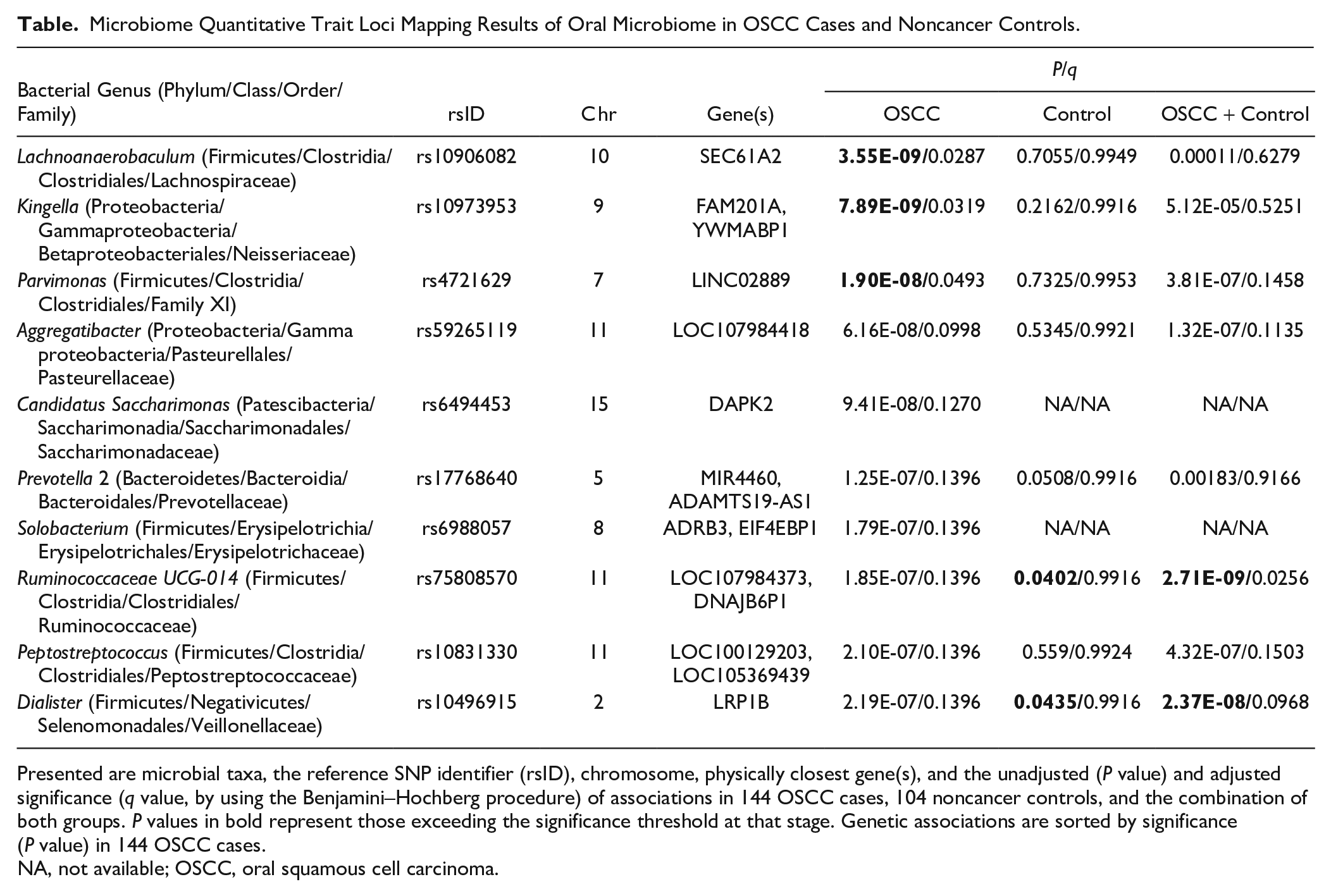
Presented are microbial taxa, the reference SNP identifier (rsID), chromosome, physically closest gene(s), and the unadjusted (P value) and adjusted significance (q value, by using the Benjamini–Hochberg procedure) of associations in 144 OSCC cases, 104 noncancer controls, and the combination of both groups. P values in bold represent those exceeding the significance threshold at that stage. Genetic associations are sorted by significance (P value) in 144 OSCC cases.
NA, not available; OSCC, oral squamous cell carcinoma.
In addition, to warrant an OSCC-specific link between host genotypes and oral microbiome, we further performed a targeted replication analysis on top 4 hits of taxonomic associations using a previously reported cohort of 67 OSCC patients (Lee et al. 2017). One signal between rs10906082 with the abundance of Lachnoanaerobaculum, which reached the study-level significance (P < 5.2 × 10–9) in the discovery analysis, was reproduced in the replication cohort (P < 0.05 at replication and P < 2.15 × 10–4 in the meta-analysis as adjusted for sex and age) (Fig. 3A). Mapping the association of rs10906082 with Lachnoanaerobaculum in the discovery cohort using variants prior to linkage disequilibrium (LD) pruning revealed that several SNPs in high linkage disequilibrium were located within a 50-kb region that spans 2 coding genes, SEC61A2 and NUDT5, and 1 regulatory RNA gene, MIR548AK (Fig. 3B). These Lachnoanaerobaculum-associated SNPs regulated the expression of SEC61A2 or NUDT5 in multiple human tissues as determined by expression quantitative trait locus (eQTL) studies (GTEx database) (Appendix Fig. 1). By calculating the proportion of oral dysbiosis (58 genera tested) explained by host genetic variations in the discovery cohort, despite a lack of statistic power in the inference due to a limited sample size, Lachnoanaerobaculum remained 1 of 14 genera exhibiting nonzero estimates (data not shown), suggesting a genetic impact on the abundance of oral Lachnoanaerobaculum in OSCC. Moreover, our targeted analysis verified another 2 associations (Appendix Table 3), rs10973953 with the abundance of Kingella (P = 1.38 × 10–9) and rs4721629 with the abundance of Parvimonas (P = 3.53 × 10–8) in the meta-analysis as adjusted for sex and age, although not reproducible in the replication (P > 0.05). These findings suggest a link between host genetics and oral microbial composition in OSCC patients across independent cohorts.
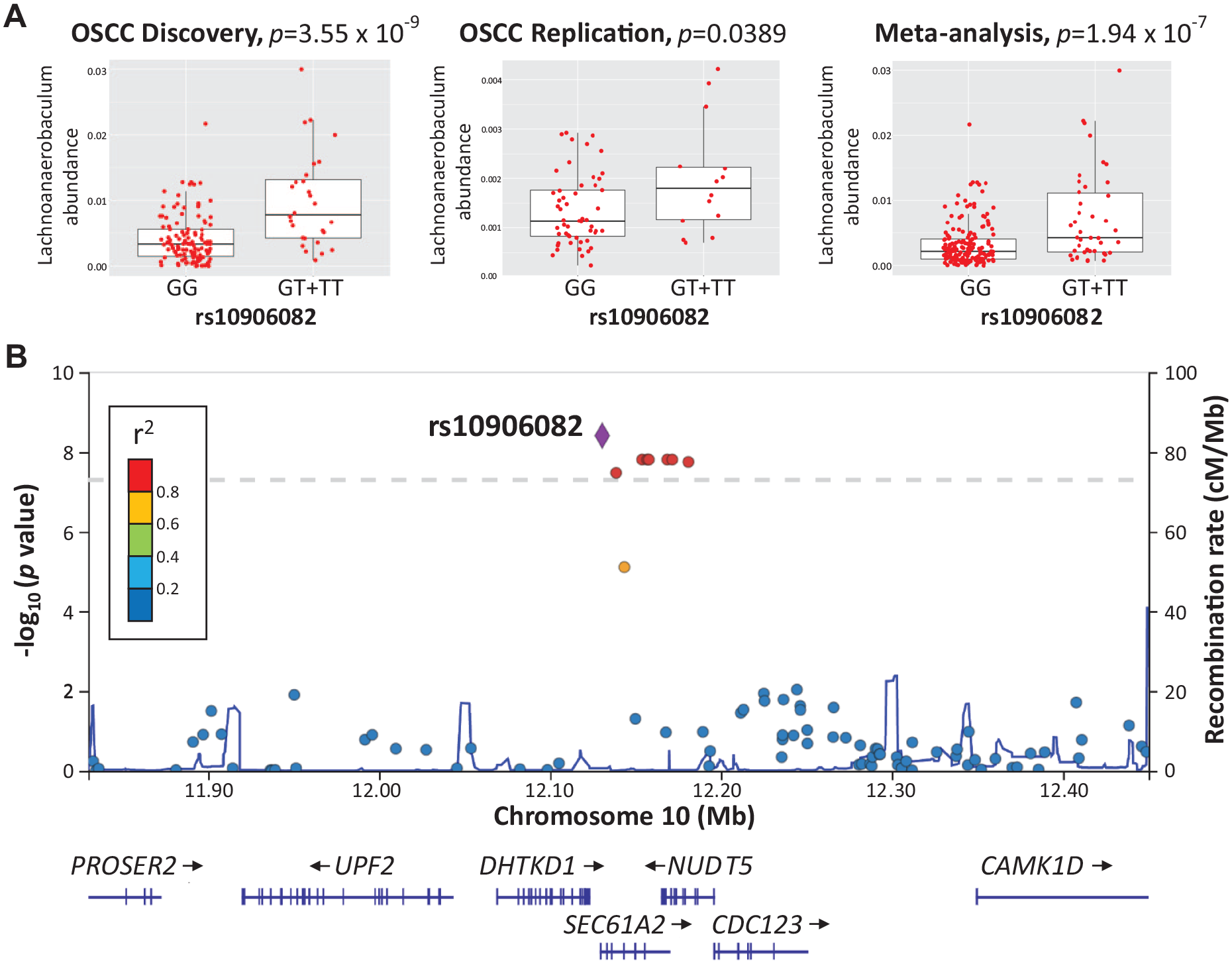
Association of microbiome quantitative trait loci (rs10906082) with oral Lachnoanaerobaculum abundance in oral squamous cell carcinoma (OSCC). (
Host Carcinogenic Gene Pathways Linked to the Oral Microbiome in OSCC
To understand how host inherited factors functionally interacted with the oral microbiome in OSCC, we performed a functional enrichment analysis on 2 gene sets identified to be associated with all oral microbial traits (diversity and taxonomy) or only taxonomy at q < 0.2 using IPA (Appendix Table 4). The most significant “functions annotation” found was “formation of solid tumor” for both gene sets (P = 8.51 × 10–8, 80 of 87 diversity and taxonomy-associated genes involved;P = 8.38 × 10–6, 55 of 60 taxonomy-associated genes involved). We further generated interconnected gene networks using both gene sets based on a curated database of biological activities and functional commonalities within the IPA. Using 87 diversity- and taxonomy-associated genes, a top-scored network returned was enriched for “growth of tumor” (P = 0.0012, 16 genes) and comprised many hubs involved in oncogenic signaling, including ERK1/2, Creb, and TGFB2 (Fig. 4A). We found that the “diseases annotation” for another network with a score of 33, centered on NF-κB complex, P38 MAPK, and PI3K complex, involved many cancer types, including “head and neck squamous cell carcinoma” (P = 0.0044, 10 genes) (Fig. 4B). Of note, this cancer-related network contained a number of metalloproteinases or their substrates (ADAMTS3, ADAMTS19, and KIRREL3), as they represent key regulators of the tumor microenvironment (Kessenbrock et al. 2010). In addition, the highest-scored network returned from the use of 60 taxonomy-associated genes was enriched for “migration of endothelial cell” (P = 0.00042, 10 genes) and consisted of several hubs implicated in angiogenesis, such as ERK1/2, Mapk, and Vegf (Appendix Fig. 2). These results provide functional insights into host genetic influences on shaping the structure of the OSCC microbiome.
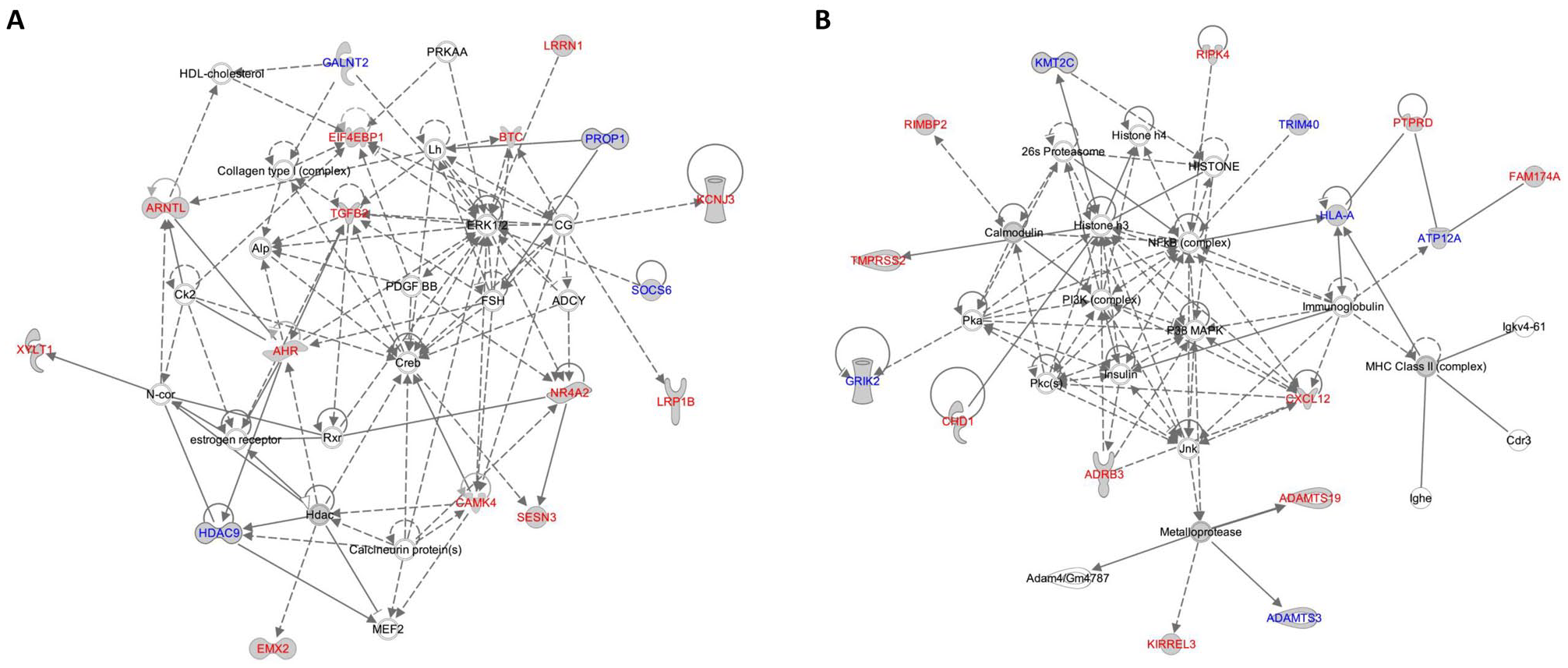
Ingenuity Pathway Analysis (IPA) networks of oral squamous cell carcinoma (OSCC)–related microbiome quantitative trait loci (mQTL) genes. Two top-scored networks of genes near mQTLs in OSCC are presented. (
Discussion
Dysbiosis of the oral microbiome in mouth cancers is nominally attributed by many factors. Yet, to date, how and to what extent host genetics govern the adaption of the OSCC microbiome in a such unique ecosystem remain unknown. We have previously delineated a relationship between the oral microbiome and somatic mutations in the tumors of the oral cavity (Yang et al. 2018). Here, a complex interaction between host germline genetic variations and oral dysbiosis in OSCC patients was reported. Among these, the genome-wide significant association between rs10906082 and the genus Lachnoanaerobaculum was the strongest (P = 3.55 × 10–9) in our study and replicated at the same allelic direction in a second OSCC cohort. A significant difference in the relative abundance of oral Lachnoanaerobaculum has been observed between OSCC patients who neither consumed betel quid nor smoked cigarettes and noncancer controls (Lee et al. 2017). Lachnoanaerobaculum belongs to the family of Lachnospiraceae, a taxon whose abundance in keratinized gingiva has been found to be influenced by host genetic variations (Blekhman et al. 2015). This locus includes SNPs located in 2 coding genes, SEC61A2 and NUDT5, and a small regulatory RNA gene, MIR548AK. The product of the SEC61A2 gene is a component of a channel-forming translocon complex that can be translocated from the membrane of the endoplasmic reticulum toward antigen-containing endosomes under the regulation mediated by Toll-like receptor (TLR) ligands, thus being essential for antigen translocation and cross-presentation (Zehner et al. 2015). The NUDT5 gene, encoding a Nudix hydrase that eliminates toxic nucleotide derivatives from the cell to ensure accurate DNA replication (Ishibashi et al. 2003), has been implicated in the response of a hypoxia-activated prodrug, evofosfamide, for treating head and neck cancers (Jamieson et al. 2018). This mQTL also harbors a member (MIR548AK) of a recently discovered family of human miRNA genes, hsa-miR-548. MicroRNA-548, whose putative target genes were identified to play cancer-related regulatory roles (Piriyapongsa and Jordan 2007), has been demonstrated to downregulate host antimicrobial response via directly targeting to interferon expression (Li et al. 2013). These observations suggest that mQTLs overlap complex disease-linked genes, unveiling a mechanistic role of these genotype–microbe interactions in OSCC pathogenesis. In addition, 2 members (Aggregatibacter and Kingella) of the HACEK microbes, a group of fastidious, Gram-negative oropharyngeal commensals that have been correlated with the etiology of periodontitis and other infections (Norskov-Lauritsen 2014), were identified among top association signals (rs10973953 with Kingella, P = 7.89 × 10–9; rs59265119 with Aggregatibacter, P = 6.16 × 10–8). Disturbance in salivary levels of Aggregatibacter has been detected in oral cancer (Furquim et al. 2017; Guerrero-Preston et al. 2016), and this taxon in supragingival plaque was shown to be heritable (Blekhman et al. 2015). Different from the mQTL overlapped to the STAB1 and ESRRA gene in supragingival plaque (Blekhman et al. 2015), the Aggregatibacter-associated locus (rs59265119) identified in our study is nearby (downstream of) the NAV2 gene, which encodes for a cell migration molecule with prognostic applications in many tumor types (Ishiguro et al. 2002; Hu et al. 2019). These data suggest that host genetic effects on a given heritable taxon in the mouth are influenced by oral health and disease.
Apart from the comparisons with other oral microbiome GWASs, a universal genetic effect on human microbiota colonized in multiple habitats remains an open question. The SLIT3 gene was previously demonstrated to harbor many variants in association with the gut microbiome in TwinsUK (Goodrich et al. 2016) and genetically unrelated subjects (Bonder et al. 2016), as well as with airway microbes in the Hutterites (Igartua et al. 2017). A suggestive interaction of SLIT3 gene polymorphisms with the oral microbiome in unrelated OSCC cases was also seen in our study (Appendix Fig. 2), with a hit between rs871097 and the abundance of genus Filifactor (P = 6.33 × 10–7, q = 0.1634). Silencing of the SLIT3 gene has been commonly observed in most human cancers due to frequent hypermethylation of promoters (Dickinson et al. 2004; Narayan et al. 2006; Nones et al. 2014), and its expression was augmented to regulate cell motility in macrophages in response to lipopolysaccharides (LPSs) (Tanno et al. 2007). These associations of SLIT3 SNPs detected in other mGWAS studies and in our cohort suggest that this gene may play a role in governing bacterial abundances in a genotype-specific manner across distinct human body sites.
By characterizing specific functional gene pathways in the human genome that shape the composition of the oral microbiome, we revealed that several oncogenesis- and angiogenesis-related pathways were enriched among OSCC microbiome-correlated host genes, such as Creb, NF-κB complex, PI3K, TGFB2, vegf, and MAPK (ERK1/2 and P38). This leads to a conjecture that the link between human genetics and OSCC microbiome in the oral cavity can dictate dysbiotic inclination that might predispose to oral malignancies and could be subject to intervention. Such complex relationship was noted in healthy individuals and subjects with inflammatory bowel disease (IBD) as well. In healthy populations, heritable microbiome traits were found to interact with host metabolic functions in the gut (Blekhman et al. 2015; Bonder et al. 2016) and innate mucosal immunity in the upper airway (Igartua et al. 2017). Intriguingly, in patients with IBD across independent cohorts, genes affecting gut microbial compositions were shown to be enriched for the JAK-STAT signaling in regulation of host innate immunity (Knights et al. 2014). In addition, it is noteworthy that our network construction exhibited a hub comprising multiple metalloproteinases or their substrates. This observation is in concordance with a proposed mechanism of oral carcinogenesis induced by dysbiosis (La Rosa et al. 2020), through which LPSs and bacteriocins can stimulate host cells to release collagenases, prostaglandins, and thromboxane, further sustaining mucosal damage or potentiating the release of TGFB2 to cause abnormal proliferation in oral cells. Our results indicate that functional human genomic data connected to mouth microbiota are useful for making assumptions on microbiome seeding in the oral cavity and its influence on the development of disease phenotypes.
Moreover, 2 top hits of mQTLs (rs75808570 with the genus Ruminococcaceae UCG-014 and rs10496915 with the genus Dialister) identified in OSCC cases at the discovery stage were verified in noncancer controls. Ruminococcaceae UCG-014 is assigned to the family of Ruminococcaceae, a bacterial taxon whose levels in saliva have been shown to be affected by host genetic variations (Blekhman et al. 2015). The locus linked to oral Dialister abundancies is located in the LRP1B gene, encoding a transmembrane receptor (low-density lipoprotein receptor–related protein 1B) implicated in lipoprotein metabolism. Genome-wide association analysis of obesity-related traits has reported LRP1B as a susceptibility factor in obesity (Akiyama et al. 2017). Successful verification of this genotype-microbe interaction may be, in part, accounted for by this metabolic function. In addition, LRP1B was proposed as a tumor suppressor due to its complex interactions with multiple ligands (Liu et al. 2001). This, again, reflects that certain taxonomical associations driven by genetic effects detected in our study may be not only restricted to OSCC but also relevant to the metabolic responses of healthy individuals.
Despite novel insights into host genetic influences on the OSCC microbiome provided here, extra work is needed to address several limitations of the present study. One issue is that the size of our cohort is relatively small (144 subjects in the identification; 248 and 211 subjects in the combination) for mQTL mapping and reliable estimation of heritability. Considering successful identification and replication of mQTLs, we acknowledge that many more associations are presumably to be found in a larger sample size. Another limitation is the multiple testing burden derived from the high dimensionality of oral microbiome data. As we decreased the number of tests by analyzing only genus-level taxa present in the majority of samples or bacterial diversity measures, we only corrected for sex, age, disease status, and multiple testing within each study but did not correct for recognized or region-specific etiological factors of OSCC. These etiological factors (e.g., betel quid chewing, cigarette smoking, oral hygiene, and periodontal condition) are known to influence oral microbiome and, to some extent, positively or negatively affect the individual genotype–microbe association (Appendix Table 5). However, the impact of those risks on the microbe–genotype–cancer relationship may be underestimated due to a lack of a quantitative definition for betel quid chewing, alcohol consumption, and smoking. Overall, through a similar rationale to that revealed by the observations of GWASs, the underlying biological and pathogenic processes of our association signals may surpass the statistical estimates. Taken together, our findings unveil a complex set of associations between the OSCC microbiome and host genetic variation on oncogenesis- and angiogenesis-related pathways, providing a starting point toward interrogating the host genome–microbe interaction in the context of oral tumorigenesis.
Author Contributions
S.F. Yang, contributed to conception, design, data acquisition, and interpretation, drafted the manuscript; C.W. Lin, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; C.Y. Chuang, Y.C. Lee, contributed to data acquisition and analysis, critically revised the manuscript; W.H. Chung, H.C. Lai, contributed to data interpretation, critically revised the manuscript; L.C. Chang, contributed to design and data analysis, drafted the manuscript; S.C. Su, contributed to conception, design, data acquisition, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345211051967 – Supplemental material for Host Genetic Associations with Salivary Microbiome in Oral Cancer
Supplemental material, sj-docx-1-jdr-10.1177_00220345211051967 for Host Genetic Associations with Salivary Microbiome in Oral Cancer by S.F. Yang, C.W. Lin, C.Y. Chuang, Y.C. Lee, W.H. Chung, H.C. Lai, L.C. Chang and S.C. Su in Journal of Dental Research
Footnotes
Acknowledgements
We thank Rhian Resnick and Skyler Paulus for their aid with the use of high-performance computing (HPC) service at Florida Atlantic University (FAU). We are also grateful to the Tissue Bank at Chang Gung Memorial Hospital, Keelung, Taiwan, for preparing clinical specimens.
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by research grants from Health and Welfare Surcharge of Tobacco Products, Ministry of Health and Welfare, Taiwan (MOHW109-TDU-B-212-134025, MOHW110-TDU-B- 212-144025); from Chung Shan Medical University Hospital, Taiwan (CSH-2021-E-002-Y3) to S.F. Yang; and from Chang Gung Memorial Hospital (BMRPE97) to S.C. Su.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.