
Abstract
Amelogenesis imperfecta (AI) is a diverse group of inherited diseases featured by various presentations of enamel malformations that are caused by disturbances at different stages of enamel formation. While hypoplastic AI suggests a thickness defect of enamel resulting from aberrations during the secretory stage of amelogenesis, hypomaturation AI indicates a deficiency of enamel mineralization and hardness established at the maturation stage. Mutations in ENAM, which encodes the largest enamel matrix protein, enamelin, have been demonstrated to cause generalized or local hypoplastic AI. Here, we characterized 2 AI families with disparate hypoplastic and hypomaturation enamel defects and identified 2 distinct indel mutations at the same location of ENAM, c588+1del and c.588+1dup. Minigene splicing assays demonstrated that they caused frameshifts and truncation of ENAM proteins, p.Asn197Ilefs*81 and p.Asn197Glufs*25, respectively. In situ hybridization of Enam on mouse mandibular incisors confirmed its restricted expression in secretory stage ameloblasts and suggested an indirect pathogenic mechanism underlying hypomaturation AI. In silico analyses indicated that these 2 truncated ENAMs might form amyloid structures and cause protein aggregation with themselves and with wild-type protein through the added aberrant region at their C-termini. Consistently, protein secretion assays demonstrated that the truncated proteins cannot be properly secreted and impede secretion of wild-type ENAM. Moreover, compared to the wild-type, overexpression of the mutant proteins significantly increased endoplasmic reticulum stress and upregulated the expression of unfolded protein response (UPR)–related genes and TNFRSF10B, a UPR-controlled proapoptotic gene. Caspase, terminal deoxynucleotidyl transferase UTP nick-end labeling (TUNEL), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays further revealed that both truncated proteins, especially p.Asn197Ilefs*81, induced cell apoptosis and decreased cell survival, suggesting that the 2 ENAM mutations cause AI through ameloblast cell pathology and death rather than through a simple loss of function. This study demonstrates that an ENAM mutation can lead to generalized hypomaturation enamel defects and suggests proteinopathy as a potential pathogenesis for ENAM-associated AI.
Keywords
Introduction
Dental enamel is formed through a developmental process, amelogenesis, orchestrated by ameloblasts and other enamel organ cells that determines the final shape of a tooth crown (Simmer et al. 2010). Amelogenesis is generally divided into 4 stages: presecretory, secretory, transition, and maturation (Smith and Nanci 1995; Hu et al. 2007). Following morphodifferentiation of the enamel organ, the inner enamel epithelium begins to reorganize its cellular structure and differentiate into functional ameloblasts, which constitutes the presecretory stage. Later, during the secretory stage, ameloblasts actively produce enamel matrix proteins (EMPs), primarily amelogenin (AMEL), ameloblastin (AMBN), and enamelin (ENAM), and enamel mineral ribbons elongate (appositional growth) as ameloblasts retreat, which determines the enamel thickness of a tooth (Fincham et al. 1999). Due to their heavy load of protein secretion, secretory stage ameloblasts have been shown to implement the unfolded protein response (UPR) to manage the increased endoplasmic reticulum stress (ERS) in a prosurvival mode (Tsuchiya et al. 2008), but nevertheless about 25% of ameloblasts undergo apoptosis during the following brief transition stage (Smith and Warshawsky 1977) when ameloblasts drastically diminish EMP production and cease retreating. Finally, the enamel matures (hardens) as EMPs are degraded by proteases and reabsorbed by maturation stage ameloblasts, which allows mineral deposition and thickening of enamel ribbons that ultimately interlock with one another (Smith 1998). The hardness of dental enamel is mainly established at this stage. As these cellular and chemical events must be well coordinated, any disturbance during sequential stages of amelogenesis can cause various forms of developmental enamel defects (Lacruz et al. 2017).
Amelogenesis imperfecta (AI) is a heterogenous set of genetic disorders characterized by enamel malformations (Witkop 1988). In general, AI can be phenotypically grouped into 3 categories: hypoplastic, hypomaturation, and hypocalcified (Hu et al. 2007). Hypoplastic AI defines aberrantly thin but properly mineralized enamel that is caused by disruptions in appositional growth during the secretory stage of enamel formation. In contrast, hypomaturation AI refers to hardness defects of enamel with relatively normal thickness, which results from disturbed enamel mineralization at the maturation stage. Hypocalcified AI is a special form of AI featured by markedly soft enamel that is prone to failure after tooth eruption. Genetic studies have identified many causative genes responsible for these various types of AI and revealed their roles at sequential stages of amelogenesis (Smith et al. 2017). ENAM (OMIM *606585) is an evolutionarily conserved gene that encodes an EMP, enamelin, and belongs to the secretory calcium-binding phosphoprotein (SCPP) gene family critical for biomineralization (Hu et al. 1997; Kawasaki and Weiss 2008). However, it is pseudogenized in species that do not make dental enamel, suggesting its only critical function is in enamel formation (Meredith et al. 2009). Consistently, the enamelin gene is specifically expressed by secretory stage ameloblasts of species producing enamel, including mice (Hu, Sun, et al. 2001; Gasse and Sire 2015). Mutations in ENAM have been shown to cause generalized or local hypoplastic AI, which further demonstrates its essential roles during the secretory stage of amelogenesis (Rajpar et al. 2001; Hu and Yamakoshi 2003). Enamelin is the largest EMP produced by ameloblasts and is immediately processed upon secretion by the secretory stage enamel protease, MMP20 (Yamakoshi et al. 2006). Enamelin cleavage products are proposed to be a critical component of the mineralization front apparatus at the distal membrane of ameloblasts, which shapes, elongates, and orients mineral ribbons of enamel (Simmer et al. 2021). Accordingly, in Enam null mice, no enamel crystals are formed (Hu et al. 2008; Smith et al. 2016).
To date, more than 20 different ENAM mutations have been reported in human AI kindreds, all of which are associated with hypoplastic enamel defects (Yu et al. 2022). In this study, we identified 2 genetic variants at the same location of ENAM that distinctively caused hypoplastic and hypomaturation AI. Further analyses indicated that the malformed enamel likely results from disturbed ameloblast secretion, increased ERS and UPR, and subsequent cell apoptosis. The results not only expand the phenotypic and genotypic spectrums of ENAM mutations but also reveal potential pathogenic mechanism of ENAM-associated AI.
Materials and Methods
Subject Recruitment and Mutational Analyses
The protocols for human research and consent forms were reviewed and approved by the institutional review board committee at the National Taiwan University Hospital. All recruitment procedures and study processes were detailed in our protocols and complied with the Declaration of Helsinki. All participants signed written consents after being given a thorough explanation and discussion about the research procedures. After history taking, orodental examinations were performed for phenotyping and constructing the family pedigree. A 2-mL sample of unstimulated saliva was obtained from each subject and used to obtain genomic DNA for subsequent genetic analyses.
Whole exome sequencing was conducted for the proband of each family using the KAPA HyperCap target enrichment system (Roche) coupled with Novaseq 6000 sequencing platform (Illumina), followed by bioinformatic analysis as previously described (Wang et al. 2019). Sanger sequencing was performed to validate identified sequence variants and assess their segregation with disease phenotypes. The American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines were employed to assess, categorize, and interpret the sequence variants (Richards et al. 2015). The mutations were annotated and described based on human reference sequences, NG_013024.1, NM_031889.3, and NP_114095.2. The exons were numbered by counting in exon 2, which is not included in humans (NM_031889.3) but in mice (NM_017468.3), to keep the numbering consistent with precedent publications (Hu et al. 2000; Hu, Zhang, et al. 2001).
In Situ Hybridization
Research protocols and procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Michigan. This study used 1 C57BL/6j mouse and followed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Following tissue processing and sectioning, the RNAscope 2.5 HD (RED) Assay (Advanced Cell Diagnostics) was performed on longitudinal sections of day 14 mouse mandibular incisors using an antisense Enam riboprobe, Mm-Enam (cat. #564301, targeting NM_017468.3, nt 1128-2174), and a negative control probe, Neg Ctrl Probe_dapB (cat. #310043). The detailed procedures of tissue processing, staining, and imaging were described previously (Liang et al. 2021).
In Silico Analyses of ENAM Proteins
The PASTA 2.0 (Prediction of Amyloid STructure Aggregation 2.0) server (Walsh et al. 2014) was employed to analyze amino acid sequences of the human wild-type (WT) (NP_114095.2) and 2 truncated ENAM proteins, p.Asn197Ilefs*81 (T81) and p.Asn197Glufs*25 (T25). Three sequences of 1,103 (WT, Met40-Ala1142), 237 (T81, Met40-Asp276), and 181 (T25, Met40-Val220) amino acids, which excluded the enamelin 39-amino-acid signal peptide sequence, were used as input. The protein–protein mode was chosen to evaluate aggregations between protein homodimers and heterodimers. The top 20 best energy pairings were selected, and the energy cutoff was set to be −5.0 PASTA Energy Unit (PEU; 1.0 PEU = 1.192 Kcal/mol).
Functional Investigations of ENAM Mutations and Proteins
Experimental procedures of plasmid transfection, immunoblotting, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR), minigene, secretion, terminal deoxynucleotidyl transferase UTP nick-end labeling (TUNEL), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays are described in Appendix Materials and Methods.
Results
Family 1
The proband of family 1 (III:1) was a 5.5-y-old boy who had hypoplastic AI but was otherwise healthy (Fig. 1A). The family pedigree indicated a dominant pattern of disease inheritance from the maternal side. Clinically, he had a widely spaced primary dentition in which the teeth were somewhat microdontic and showed only a thin layer of dental enamel (Fig. 1B). Dental attrition was evident, especially over the occlusal surface of the molars. The remaining enamel looked opaque and hypomineralized. Radiographically, the enamel of all his primary and developing permanent teeth appeared severely hypoplastic while showing relatively normal contrast with underlying dentin (Fig. 1C). The proband’s mother (II:4), uncle (II:5), and maternal grandmother (I:4) all had similar enamel defects as his, suggesting a consistent phenotypic expressivity.
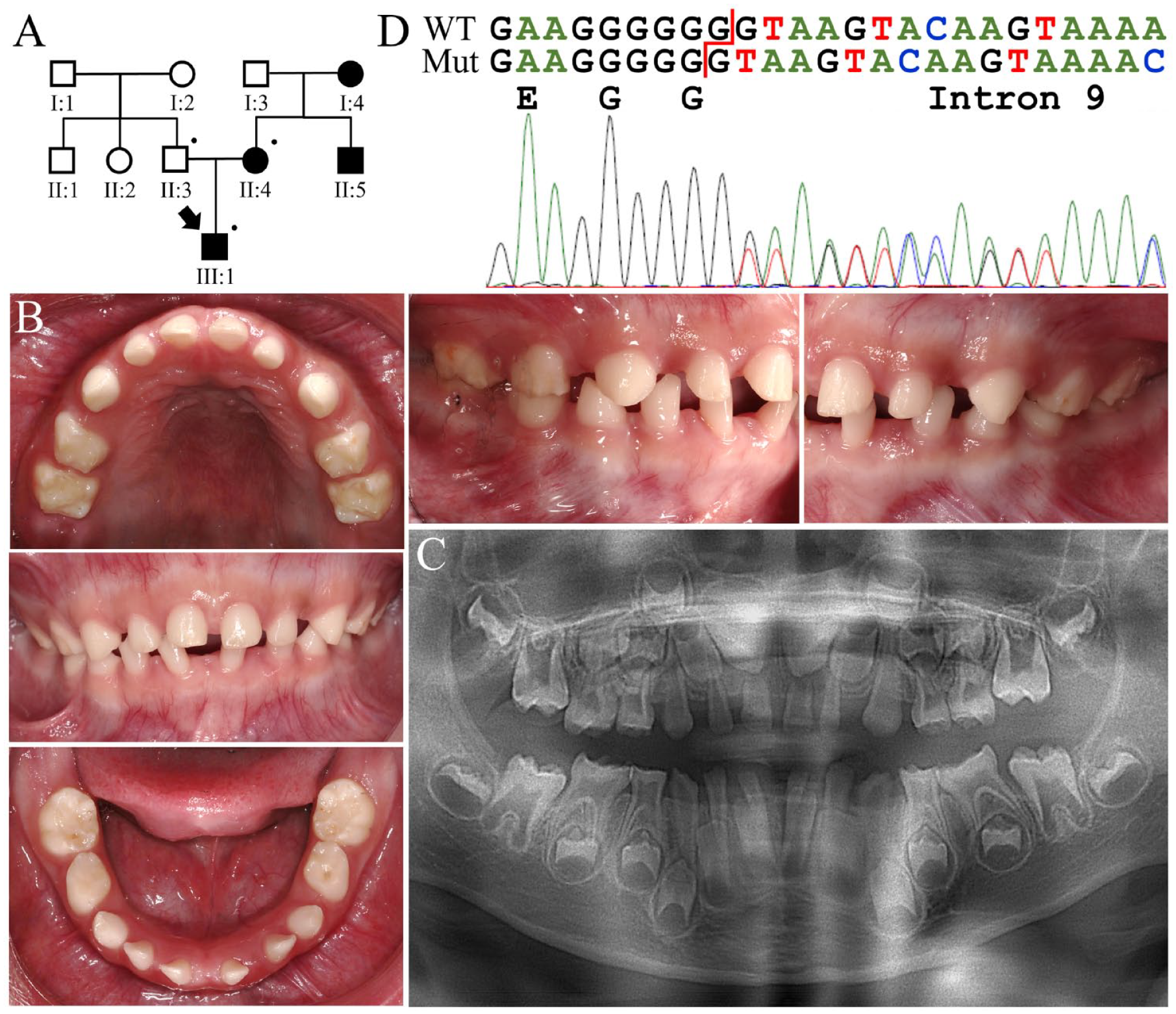
Family 1 with a heterozygous ENAM c.588+1del mutation causing generalized hypoplastic amelogenesis imperfecta. (
Exome analysis of the proband’s DNA identified a heterozygous single-nucleotide deletion in ENAM (g.14101del; c.588+1del), which has previously been reported to cause hypoplastic AI (ACMG classification: pathogenic, PVS1 + PS3 + PM2 + PP1) (Fig. 1D) (Kida et al. 2002). The deleted nucleotide is a guanine (G) located at a repeat of 7 Gs over the junction of exon 9 and intron 9 of ENAM and predicted to cause a −1 frameshift and protein truncation (p.Asn197Ilefs*81) rather than altering normal messenger RNA (mRNA) splicing. Further analyses indicated that the affected mother was also heterozygous for the ENAM mutation but not the unaffected father.
Family 2
Family 2 was a 3-generation family in which at least 5 individuals were affected with AI (Fig. 2A). The family pedigree suggested that the enamel malformation was inherited in an autosomal dominant manner, although the disease phenotype could not be confirmed in the first generation. The proband (III:6), a 12-y-old male, was generally healthy and had no history of significant perinatal or childhood illness. Clinically, all his permanent teeth appeared chalky white or slightly yellowish, without normal glaziness and translucency (Fig. 2B). The enamel did not look particularly thin except for the lingual surfaces of mandibular anterior teeth. Consistent with the clinical findings, the panoramic radiograph showed that his enamel was generally of normal thickness but less radiopaque, indicating a hypomineralized, rather than hypoplastic, enamel defect (Fig. 2C). Reviewing his dental records from the early and late mixed dentition stages further confirmed the diagnosis of hypomaturation AI (Appendix Fig. 1). His father presented a similar enamel phenotype and had undergone extensive dental treatment (Appendix Fig. 2).
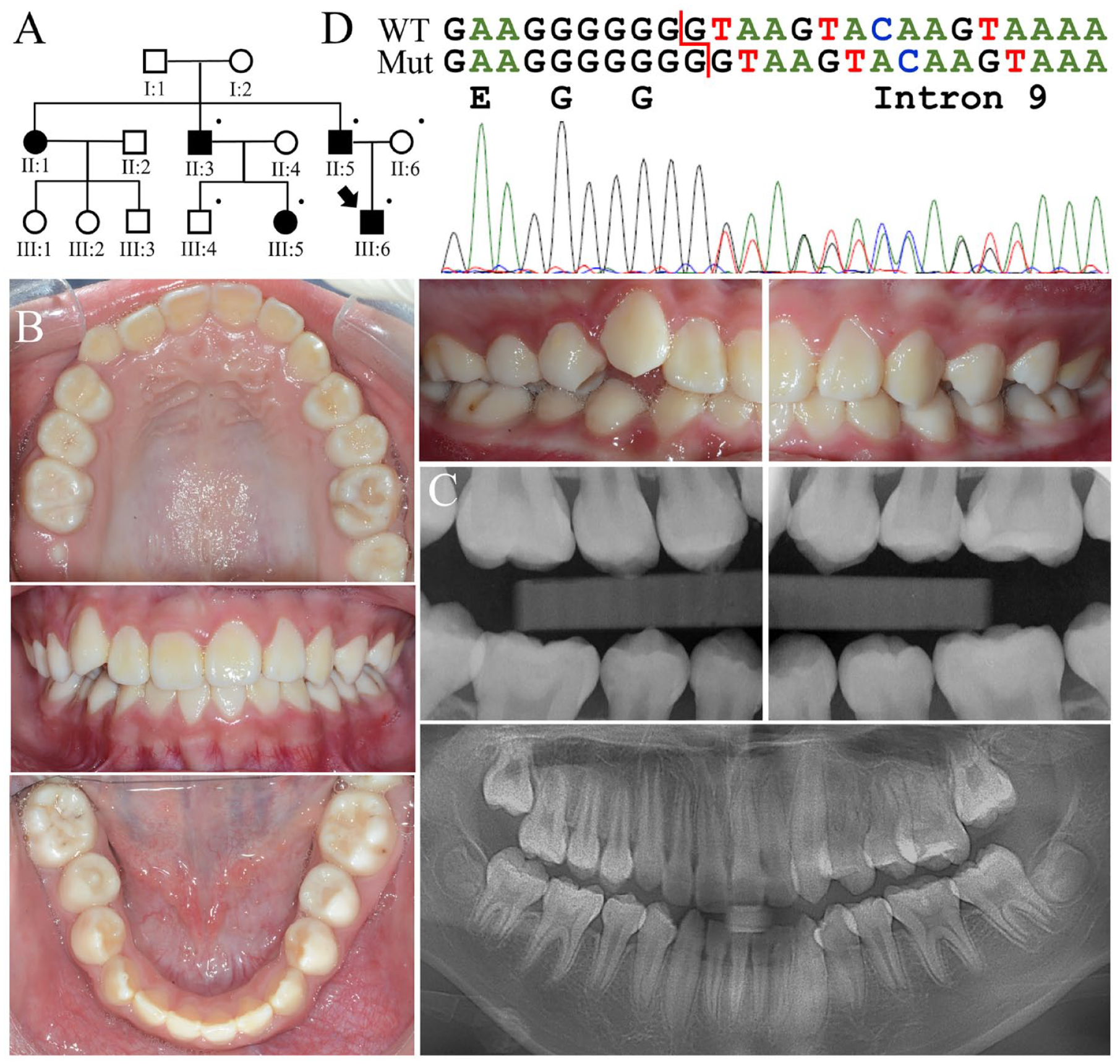
Family 2 with a heterozygous ENAM c.588+1dup mutation causing generalized hypomaturation amelogenesis imperfecta. (
Analysis of the proband’s exome revealed a single-nucleotide duplication in ENAM (g.14101dup; c.588+1dup) (Fig. 2D). The mutation is located at the same position as that of c.588+1del mutation identified in family 1. However, instead of deleting, it added a guanine to the repeat and generated a stream of 8 Gs. This sequence variant is not documented in 1000 Genomes, gnomAD, or Taiwan Biobank databases but was found by clinical testing in 3 of 264,690 chromosomes (minor allele frequency (MAF) = 0.000011) from the TOPMed database (Taliun et al. 2021). The added (eighth) G would create a new donor splice site (GT) 1 nucleotide downstream and introduce a 1-nucleotide insertion into the ENAM mRNA transcript. This would shift translation to the +1 reading frame and generate a premature termination codon in exon 10, replacing the enamelin C-terminal 946 amino acids with 24 frameshifted ones (p.Asn197Glufs*25). Target analyses demonstrated a perfect segregation between presence of the mutation and the enamel phenotype in the family, demonstrating that ENAM c.588+1dup defect causes an autosomal dominant hypomaturation AI (ACMG classification: pathogenic, PVS1 + PS3 + PM2 + PP1).
Enam Expression in Mouse Mandibular Incisors
To investigate how an ENAM mutation can cause hypomaturation AI, we reiterated Enam expression during amelogenesis using a mouse mandibular incisor, of which a longitudinal section presents all sequential stages of enamel formation. In situ hybridization with an antisense probe revealed that Enam was specifically expressed by ameloblasts but not other tissues and cells of the mandible (Fig. 3A). The signal was first detected in presecretory stage ameloblasts, continued throughout the secretory stage, and ceased abruptly following the transition stage. Only a trace amount of signal, if any, could be found in maturation stage ameloblasts. Consistently, no expression was detected in the first and second D14 molars, which contain maturation stage ameloblasts and reduced enamel epithelium. This result demonstrated a restricted, virtually exclusive, expression of Enam in secretory stage ameloblasts.
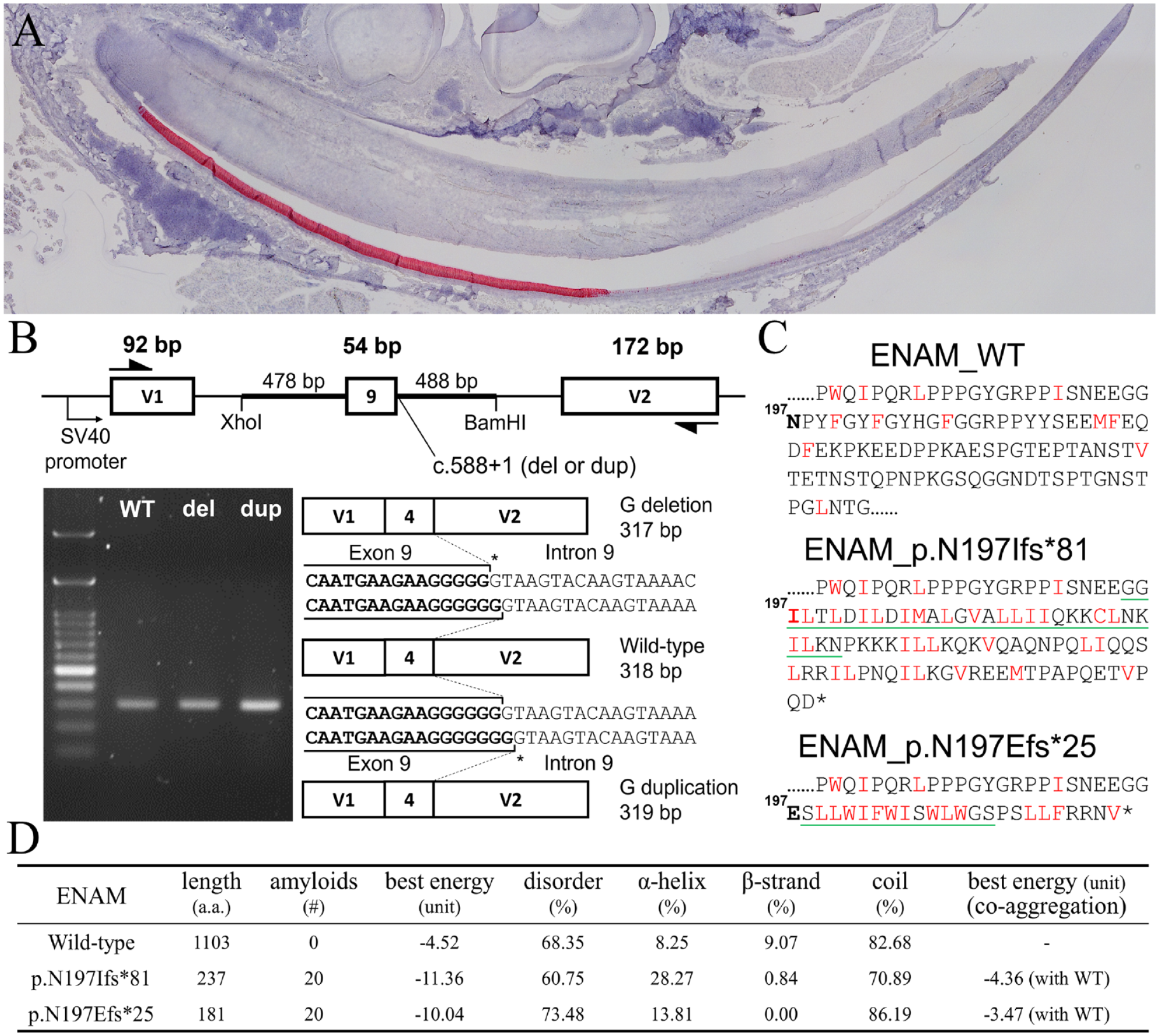
Enam expression during murine amelogenesis and analyses of human ENAM mutations. (
Molecular Characterization of ENAM Mutations and Truncated Proteins
A minigene splicing assay using the pSPL3 exon trapping vector demonstrated that the c.588+1del and c.588+1dup mutations indeed caused indels of a guanine in the open reading frame rather than abolishing the splice site (Fig. 3B). The resulting frameshifts will cause C-terminal truncation of the ENAM protein and add novel peptides of 80 and 24 amino acids, respectively, after Glycine196 (Fig. 3C). As analyzing the primary structure of the added peptides indicated a high composition of hydrophobic residues, we first performed in silico prediction of amyloid structure aggregation to see if these 2 truncated proteins, p.Asn197Ilefs*81 (T81) and p.Asn197 Glufs*25 (T25), might cause protein aggregation through hydrophobic interactions. The analyses demonstrated that, compared to the wild-type, T81 and T25 both had more predicted amyloid fibril regions, significantly decreased “the best aggregation pairing energy,” and an increased probability of aggregation (Fig. 3D). The predicted parallel aggregation region was Gly195-Asn226 for T81 and Ser198-Ser211 for T25 (Fig. 3C, Appendix Fig. 3). Furthermore, when predicted using the coaggregation mode, it revealed that T81 and T25 might aggregate with the wild-type protein with a best energy of −4.36 and −3.47 PEU, respectively (Fig. 3D). These results suggested that the truncated ENAMs might form protein aggregates with themselves and with the wild-type ENAM protein.
We therefore evaluated if the truncated proteins can be properly secreted, as aggregation might affect protein secretion. Immunoblotting demonstrated that while most of the overexpressed wild-type ENAM could be detected in the culture medium of transiently transfected HEK293T cells, both truncated proteins could be found only in cell lysates, indicating that T81 and T25 cannot be secreted properly, although their signal peptide is intact (Fig. 4A). We further conducted coexpression experiments and found that when coexpressed with T81 or T25, the wild-type ENAM was mainly detected in cell lysates rather than the culture medium, suggesting a dominant negative effect from the mutant protein on normal secretion of the wild-type ENAM (Fig. 4A).
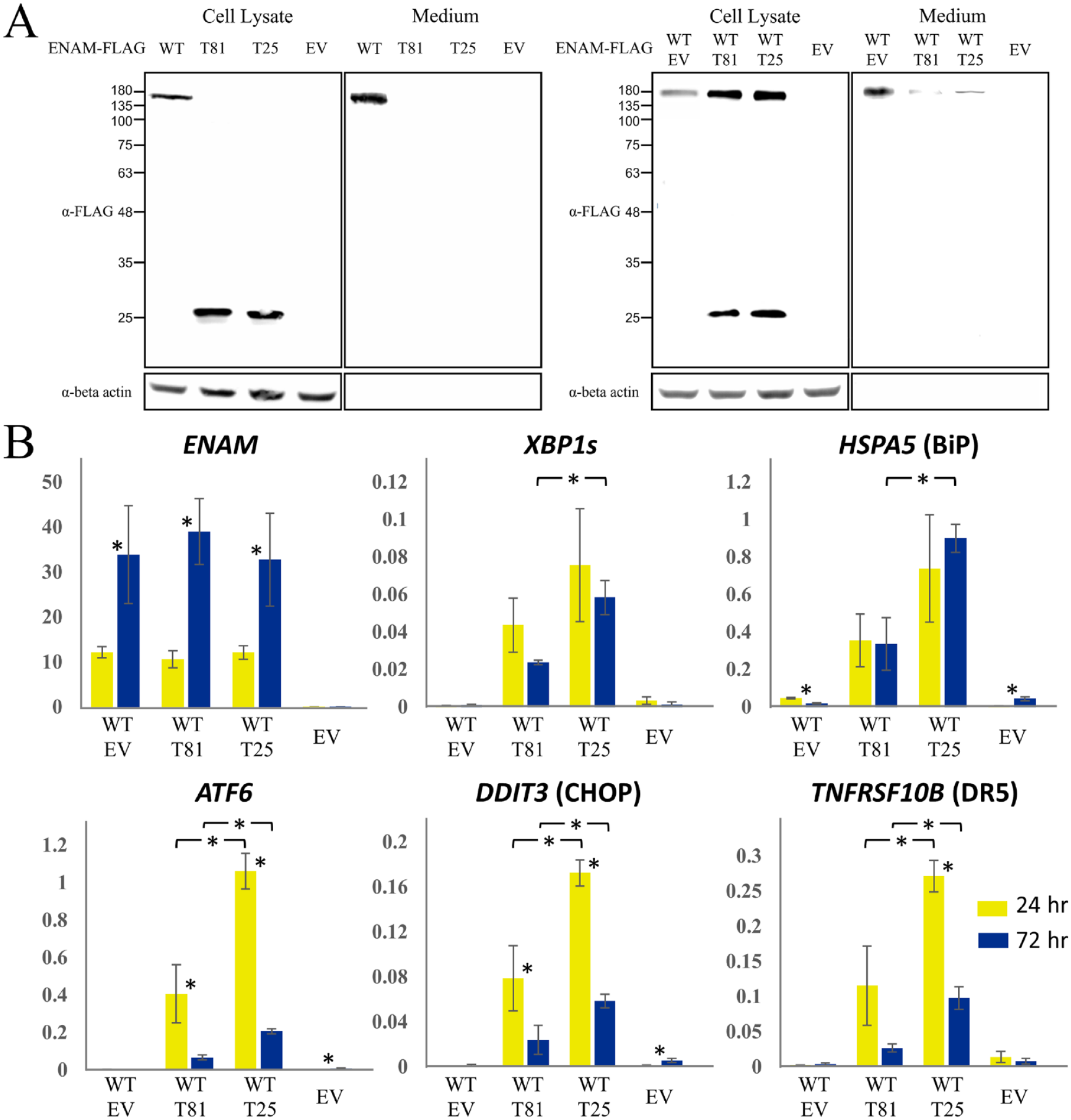
Impeded secretion and increased endoplasmic reticulum stress caused by truncated ENAM proteins. (
To investigate whether the nonsecretion of the truncated proteins causes increased ERS and potentially activates cell apoptosis, we assessed the expression of ERS-related genes, including XBP1s, HSPA5, ATF6, and DDIT3, and TNFRSF10B, a gene that encodes death receptor 5 (DR5), which promotes apoptosis under unmitigated ERS (Fig. 4B). The results showed that expressions of all 5 genes were significantly upregulated under expression of T81 or T25 compared to the wild-type and empty vector controls. In general, the T25 protein induced a higher expression level of these genes than T81. Interestingly, while ENAM expression was much higher at 72 h than that of 24 h, ATF6, DDIT3, and TNFRSF10B showed a significantly decreased expression, suggesting a potential resolution of ERS over time (Fig. 4B).
Further apoptosis assays revealed that overexpression of T81 and T25, but not wild-type ENAM or the empty vector control, triggered the cleavage of caspase 3 and caspase 7, signifying a pathway toward apoptotic cell death (Fig. 5A). Moreover, the TUNEL assay identified 100% and ~50% of cells displaying DNA fragmentation after 72 h of T81 and T25 expression, respectively, which indicated that T81 induced more pronounced cell apoptosis compared to T25 (Fig. 5B). Consistently, the MTT assays demonstrated that, while cell survival remained comparable between cells expressing wild-type ENAM and those with the empty vector control, a significantly lower number of viable cells were observed under the expression of the 2 mutant proteins (Fig. 5C). Particularly, the cell survival of T81 was lower than that of T25, suggesting that T81 is more detrimental to the cells than T25.
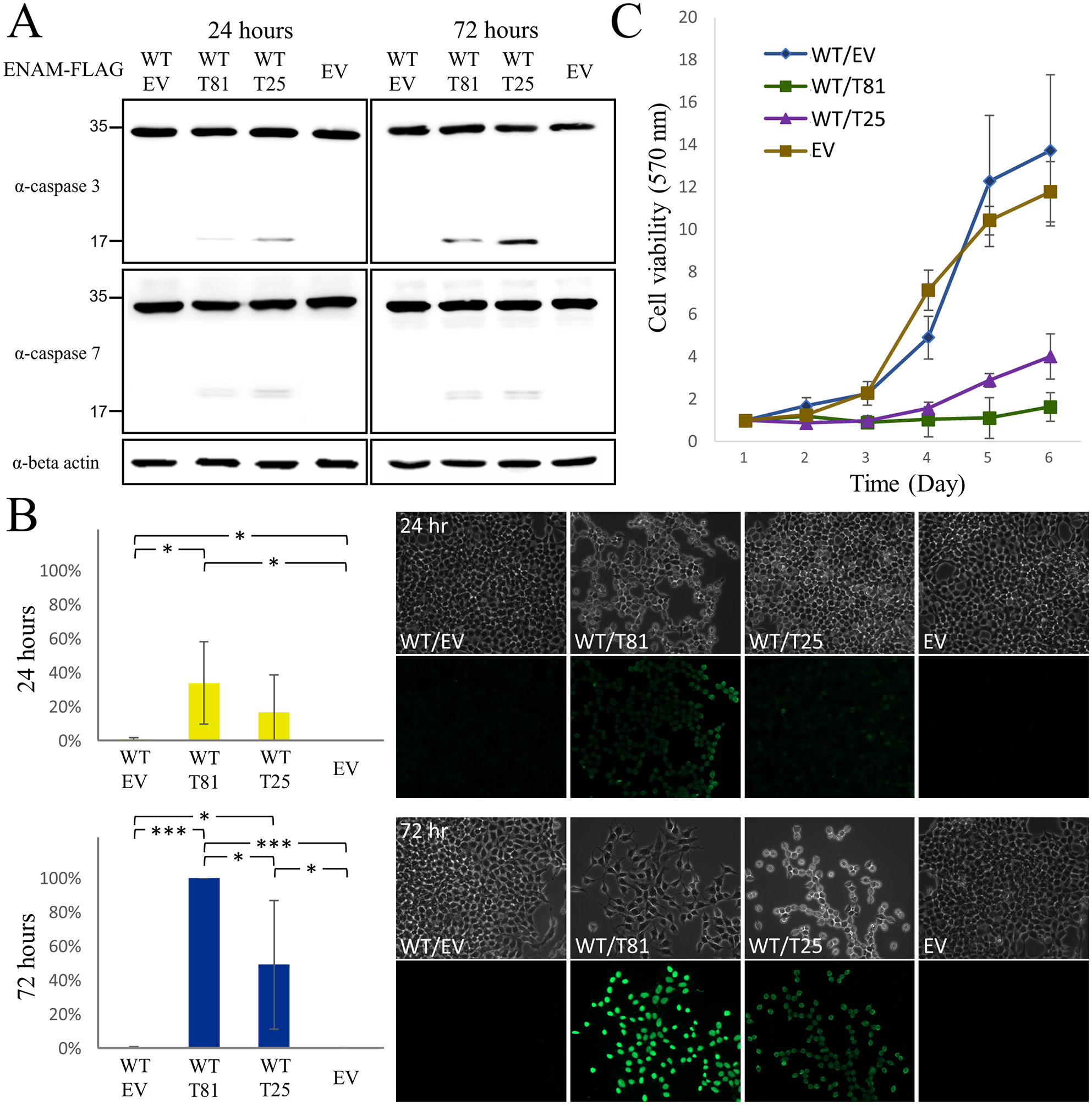
Apoptotic cell death and decreased cell survival caused by truncated ENAM proteins. (
Discussion
In this study, we identified a respective deletion and duplication mutation at the same position of ENAM, c.588+1del and c.588+1dup, that caused distinct enamel phenotypes of hypoplastic and hypomaturation AI. The deletion mutation found in family 1 has been previously reported in at least 9 AI kindreds of various ethnicities from all over the world. All of the heterozygous individuals showed generalized severe enamel hypoplasia (Zhang et al. 2019). The affected teeth usually appeared yellow due to markedly thin enamel, allowing the color of underlying dentin to show through. Although this mutation is extremely rare and not documented in public genomic databases, its high phenotypic penetrance and expressivity make it the most frequently identified variant among all the ENAM disease-causing mutations reported to date (Yu et al. 2022). In contrast, the duplication mutation identified in family 2, while being rare, is documented in the TOPMed database and designated as rs752102959 (Taliun et al. 2021). It has also been reported in the ClinVar genomic archive with 2 submissions of clinical testing (accession: VCV000427132.2) and interpreted as pathogenic (Landrum et al. 2018). However, no dental phenotypes have been detailed in these databases. Recently, this variant was found in an AI family in which the affected individuals had hypoplastic but also hypomaturation whitish enamel (Bloch-Zupan et al. 2023). Here, we showed that this mutation perfectly cosegregates with the hypomaturation enamel defects in family 2, which demonstrates the pathogenicity of the ENAM c.588+1dup mutation and, along with the recent report, confirms that an ENAM mutation can cause autosomal dominant hypomaturation AI.
The enamel phenotype associated with ENAM mutations is dose dependent (Ozdemir et al. 2005). While biallelic mutations generally cause generalized severe hypoplastic AI, heterozygous mutations usually lead to only localized enamel pits (Hart et al. 2003) or horizontal hypoplastic bands with a highly variable phenotypic expressivity and sometimes incomplete penetrance (Seymen et al. 2014; Koruyucu et al. 2018). The correlation between heterozygosity and mild severity is particularly evident for the 5′ nonsense mutations and frameshifts that likely cause the mutant transcripts to undergo nonsense mediated decay (NMD), indicating that a simple reduction in ENAM quantity (haploinsufficiency) is generally tolerated during human amelogenesis (Seymen et al. 2014). Similarly, the 3′ mutations that do not activate NMD but instead express a truncated ENAM protein, such as p.Tyr614* and p.Ser693*, generally cause localized and less pronounced enamel defects in the heterozygous carriers, suggesting a partial retention of function for the truncated proteins (Koruyucu et al. 2018; Yu et al. 2022). Unlike these cases, the 2 frameshift mutations investigated in this study, c588+1del/p.Asn197Ilefs*81 and c588+1dup/p.Asn197Glufs*25, cause generalized enamel malformations in almost all heterozygotes (Zhang et al. 2019). This high penetrance and expressivity suggest a disease mechanism other than haploinsufficiency. The protein secretion assays in this study showed that the 2 truncated ENAMs cannot be secreted properly and also hinder the normal secretion of the coexpressed wild-type protein, indicating a dominant negative effect. In silico analyses also suggested that this impediment of protein secretion probably results from protein aggregation through the added frameshifted peptide region at the C-terminus of the truncated proteins. We further demonstrated that altered ENAM secretion leads to increased ERS, activates the UPR, and induces apoptosis of transfected cells. Consistently, an AI mouse model carrying an Enam p.Ser55Ile mutation was previously shown to have enamel defects caused by obstruction of the ameloblast secretory pathway, which resulted in elevated ERS, activated UPR, and increased apoptotic ameloblasts (Brookes et al. 2017). Therefore, besides impeding normal ENAM secretion, the 2 truncated ENAM mutations likely cause ameloblast cell pathology and death, which leads to generalized severe enamel defects.
Nevertheless, how 2 alternative frameshift mutations at the same position cause distinct enamel phenotypes (hypoplastic AI for c588+1del/p.Asn197Ilefs*81 and hypomaturation AI for c588+1dup/p.Asn197Glufs*25) remains to be elucidated. Especially puzzling is how defects in ENAM, a secretory stage–specific enamel protein, cause hypomaturation enamel defects. Our in situ hybridization demonstrated strong and specific Enam expression in secretory stage ameloblasts with trace signal in maturation stage ameloblasts, which is consistent with previous findings (Hu, Sun, et al. 2001). This restricted expression pattern suggests the hypomaturation defects most likely result from pathologic effects caused by the truncated ENAM protein with a unique 24-amino-acid frameshifted C-terminus. The mutant c.588+1dup/p.Asn197Glufs*25 ENAM is expressed during the secretory stage, but the protein or its impacts persist into the subsequent maturation stage of amelogenesis. While the c.588+1del/p.Asn197Ilefs*81 frameshifted ENAM causes severe hypoplastic AI by inducing significant ERS and massive secretory stage ameloblast apoptosis, secretory ameloblasts expressing the c.588+1dup/p.Asn197Glufs*25-frameshifted ENAM might resolve the induced ERS and minimize secretory ameloblast cell death that avoids disruption of appositional growth. However, accumulation of the p.Asn197Glufs*25-frameshifted ENAM in ameloblasts after postsecretory transition could disturb the process of enamel maturation, causing the hypomaturation phenotype. It has been demonstrated that DR5 acts intracellularly as a measure to determine the persistence or resolution of ERS (Lu et al. 2014; Lam et al. 2020). The increased or decreased level of DR5 mRNA drives cells toward apoptosis or adaptation, respectively. The significant downregulation of TNFRSF10B (DR5) expression after 72 h of ENAM p.Asn197Glufs*25 overexpression compared to that of 24 h suggests that the ERS can be resolved and apoptosis prevented. The TUNEL and MTT assays, demonstrating a reduced cell apoptosis and an increased cell survival associated with p.Asn197Glufs*25 compared to p.Asn197Ilefs*81, further supports this hypothesis. However, further investigations using in vivo models are warranted to elucidate the molecular mechanism of hypomaturation enamel defects caused by the ENAM c.588+1dup mutation.
Author Contributions
Y.-L. Wang, contributed to conception, design, data acquisition and analysis, drafted and critically revised the manuscript; H.-C. Lin, S.-K. Wang, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; T. Liang, contributed to design, data acquisition and interpretation, critically revised the manuscript; J.C.-Y. Lin, J.P. Simmer, J.C.-C. Hu, contributed to conception, data analysis and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345241236695 – Supplemental material for ENAM Mutations Can Cause Hypomaturation Amelogenesis Imperfecta
Supplemental material, sj-docx-1-jdr-10.1177_00220345241236695 for ENAM Mutations Can Cause Hypomaturation Amelogenesis Imperfecta by Y.-L. Wang, H.-C. Lin, T. Liang, J.C.-Y. Lin, J.P. Simmer, J.C.-C. Hu and S.-K. Wang in Journal of Dental Research
Footnotes
Acknowledgements
We thank the families for participating in the study and the staff of the Biomedical Resource Core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance with molecular cloning and plasmid construction.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Ministry of Science and Technology in Taiwan (MOST) grants, 108-2314-B-002-038-MY3 (S.-K. Wang) and 111-2314-B-002-111-MY3 (S.-K. Wang); National Taiwan University Hospital (NTUH) grants, 112-UN0010 (S.-K. Wang) and 112-UN0048 (Y.-L. Wang); and National Institutes of Health grants, R01DE027675 (J.P. Simmer) and R56DE015846 (J.C.-C. Hu).
A supplemental appendix to this article is available online.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.