
Abstract
The regenerative capacity of dental pulp declines as dental pulp stem cells (DPSCs) enter senescence, compromising pulp–dentin homeostasis, and actionable strategies to restore odontogenesis remain limited. Here, we identify that nitrate acts as a Sialin (SLC17A5)–dependent metabolic cue that promotes dentin regeneration by rewiring mitochondrial metabolism. Across 3 senescence models (natural aging, hydrogen peroxide, and etoposide), nitrate consistently reduced senescence features and restored odontogenic capacity. Silencing Sialin abolished these benefits, establishing pathway necessity. Untargeted metabolomics with pathway enrichment revealed activation of the tricarboxylic acid and pyruvate pathways, accompanied by higher adenosine triphosphate, an increased nicotinamide adenine dinucleotide redox ratio, improved mitochondrial respiratory performance, and lowered mitochondrial reactive oxygen species; these gains were blunted by Sialin loss. Pharmacologic perturbations linked metabolism to phenotype, as electron-transport inhibition negated nitrate’s effects, whereas metabolic modulation partially restored antisenescence and pro-odontogenic outcomes. In a mouse pulp exposure model, local nitrate accelerated dentin bridge formation and tissue repair, but these effects were lost in mesenchymal Sialin conditional-knockout mice. Together, these findings establish a nitrate–Sialin–mitochondria axis that rejuvenates DPSCs, drives dentin regeneration, and restores pulp–dentin homeostasis, while supporting a practical local delivery approach with translational potential for vital pulp therapy.
Keywords
Introduction
Regenerating dentin requires a vital pulp in which dental pulp stem cells (DPSCs) retain proliferative and odontogenic capacity (Li Z et al 2025; Niu et al 2025; Zhou et al 2025). Contemporary vital pulp therapies and calcium–silicate cements achieve hemostasis, sealing, alkalinity, and limited growth factor release from dentin, yet they do not reverse DPSC senescence or rebuild a functional progenitor pool (Liu Y et al 2025; Wang Y et al 2025). Regenerative endodontic procedures can revascularize canals but frequently produce fibrodentin or bone-like tissue rather than tubular dentin, indicating incomplete control of fate under oxidative and genotoxic stress (Mei et al 2025). Mechanistically, current approaches chiefly modulate the extracellular milieu, whereas the intracellular bottlenecks that lock DPSCs in a senescent, low-odontogenic state—impaired mitochondrial respiration, redox imbalance, and a stable senescence-associated program—remain unaddressed (Wang X et al 2025). Therefore, there is a clinical need for an upstream, tractable cue that restores organelle function and resets gene-regulatory programs in senescent DPSCs, thereby enabling formation of true dentin and reestablishing pulp–dentin homeostasis (Zhang et al 2024; Yang et al 2025).
Mitochondria are central to DPSC fate decisions (Schmitz et al 2025). During mesenchymal lineage commitment, these cells undergo a glycolysis-to-oxidative phosphorylation transition that increases tricarboxylic acid (TCA) flux, raises adenosine triphosphate (ATP) output, and rebalances the nicotinamide adenine dinucleotide pool (NAD+/NADH), while constraining mitochondrial reactive oxygen species (ROS) within a narrow signaling range to maintain metabolic and redox homeostasis (Petrelli et al 2023). In parallel, mitochondrial quality control, including biogenesis, dynamics, and PINK1–PRKN–mediated mitophagy, maintains electron transport integrity and cristae architecture (Chakrabarty and Chandel 2021; Andersson et al 2025); conversely, chronic respiratory impairment or excessive mitochondrial ROS stabilizes p53–p21 and p16–RB pathways, promotes a senescence-associated secretory phenotype, and suppresses odontogenesis (Narita et al 2003; Jimenez-Loygorri et al 2024; Wei et al 2024). What remains unresolved in DPSCs is how to safely and reversibly enhance TCA/pyruvate flux and NAD+ bioavailability, optimize ROS signaling without extinguishing necessary redox cues, and demonstrate that organelle-level correction is causally sufficient to restore odontogenesis and dentin formation in vivo.
Nitrate (NO3–), traditionally framed within the nitrate–nitrite–nitric oxide (NO) pathway, is increasingly recognized as a bioactive signal that modulates vascular tone, neuronal activity, and cellular metabolism (Zhu et al 2021; Pinaffi-Langley et al 2024). The transporter Sialin (SLC17A5) concentrates nitrate in saliva and enables cellular responsiveness in oral tissues, yielding salivary levels that exceed plasma by an order of magnitude (Qin et al 2012; Rosier et al 2022; Li et al 2024; Li X et al 2025). Across mammalian systems, emerging evidence indicates that nitrate–Sialin signaling can engage compartmentalized pathways and influence mitochondrial efficiency and redox programs at physiologically achievable concentrations (Axton et al 2019; Alsharif et al 2023). Together, these observations identify nitrate as a biocompatible, inexpensive, and locally deliverable means to tune mitochondrial metabolism in stem/progenitor contexts and to help restore pulp–dentin homeostasis (Ning et al 2025). Accordingly, we test whether Sialin-dependent nitrate signaling can rejuvenate senescent DPSCs and enable dentin regeneration.
Based on these premises, we hypothesized that nitrate activates a Sialin-dependent mitochondrial program that reinstates DPSC function and tissue homeostasis. We integrated 3 complementary DPSC senescence models—natural aging, hydrogen peroxide, and etoposide—to establish the consistency of antisenescence and pro-odontogenic effects; applied Sialin loss of function to demonstrate pathway necessity; and combined untargeted metabolomics with mitochondrial readouts—ATP, NAD+/NADH, respiratory complex function, and mitochondrial ROS—to define organelle-level changes. Pharmacologic perturbations were then used to relate metabolic shifts to gains or losses in odontogenesis and senescence features. Finally, we determined Sialin dependence of dentin bridge formation in a mouse pulp exposure model. Together, these studies delineate a nitrate–Sialin–mitochondria axis that rejuvenates DPSCs and enables dentin regeneration.
Materials and Methods
Detailed descriptions of the materials and methods are presented in the Appendix.
Results
Nitrate Reduces Senescence and Restores Odontogenic Capacity across 3 Different Models of Stress in Dental Pulp Stem Cells
We verified that the dental pulp stem cells used in this study met International Society for Cell and Gene Therapy (ISCT) mesenchymal stem cell (MSC) criteria. Flow cytometry confirmed a canonical MSC immunophenotype that was positive for CD73, CD90, and CD105 and negative for CD34 and CD45 (Appendix Fig. 1A, B). In addition, these cells displayed trilineage differentiation potential under standard induction conditions (Appendix Fig. 1C). To test whether nitrate mitigates cellular senescence and preserves functional capacity, we established 3 complementary senescence models in DPSCs. Hydrogen peroxide (H2O2) and etoposide (Eto) were used to induce stress-induced premature senescence, and serial passaging to passage 15 was used to model replicative senescence. Before efficacy testing, an effective and noncytotoxic nitrate concentration was determined from dose–response analyses in the H2O2 and Eto models using senescence-associated β-galactosidase (SA-β-gal) staining and quantitative reverse transcription polymerase chain reaction (RT-qPCR) of senescence markers (Appendix Fig. 2A–L), identifying 4 mM nitrate as the working concentration used thereafter. To exclude the possibility that the observed effects reflected sodium, osmolarity, or nonspecific ionic changes, we performed an ion substitution control in the hydrogen peroxide–induced senescence model. NaNO3 and an independent nitrate source, KNO3, produced comparable antisenescence and pro-proliferative effects, whereas equimolar NaCl or KCl did not improve these readouts (Appendix Fig. 3A–G). These data support an anion-specific effect attributable to nitrate. Furthermore, to ensure that the nitrate treatment under H2O2 stress did not alter stem cell identity, we assessed mesenchymal stem cell surface markers by flow cytometry. DPSCs remained positive for CD73 and CD90 and negative for CD34 and CD45 in both the H2O2 group and the H2O2 with nitrate group (Appendix Fig. 3H–K).
Across all 3 models, nitrate treatment reduced senescence phenotypes and partially restored key functional readouts relative to untreated senescent controls. Specifically, the proportion of SA-β-gal–positive cells decreased, intracellular ROS levels declined, and senescence-associated genes—including CDKN2A, CDKN1A, and representative senescence-associated secretory phenotype (SASP) genes such as IL1A, IL1B, IL6, and LTA—were downregulated (Fig. 1A–C, I–K, Q–S). Proliferation increased, as indicated by a higher fraction of Ki67-positive cells (Fig. 1D, L, T). To assess odontogenic capacity, alkaline phosphatase (ALP) staining/activity and alizarin red S (ARS) mineral deposition were quantified; nitrate partially restored the impaired responses in senescent DPSCs (Fig. 1E–G, M–O, U–W). Consistently, senescence reduced odontogenic marker expression relative to nonsenescent controls, whereas nitrate increased the expression of odontogenic genes (DSPP, DMP1, ALPL, COL1A1, RUNX2, SPP1) and proteins (DSPP, DMP1), shifting these readouts toward control levels (Fig. 1H, P, X; Appendix Fig. 3L–N). To strengthen translational relevance, we compared dental pulp stem cells derived from younger donors (14–24 y) and older donors (55–65 y). Cells from older donors showed reduced EdU incorporation together with higher p16 expression relative to younger-donor cells (Appendix Fig. 3O, P). Nitrate treatment in older-donor cells partially reversed age-associated changes (Appendix Fig. 3O, P).
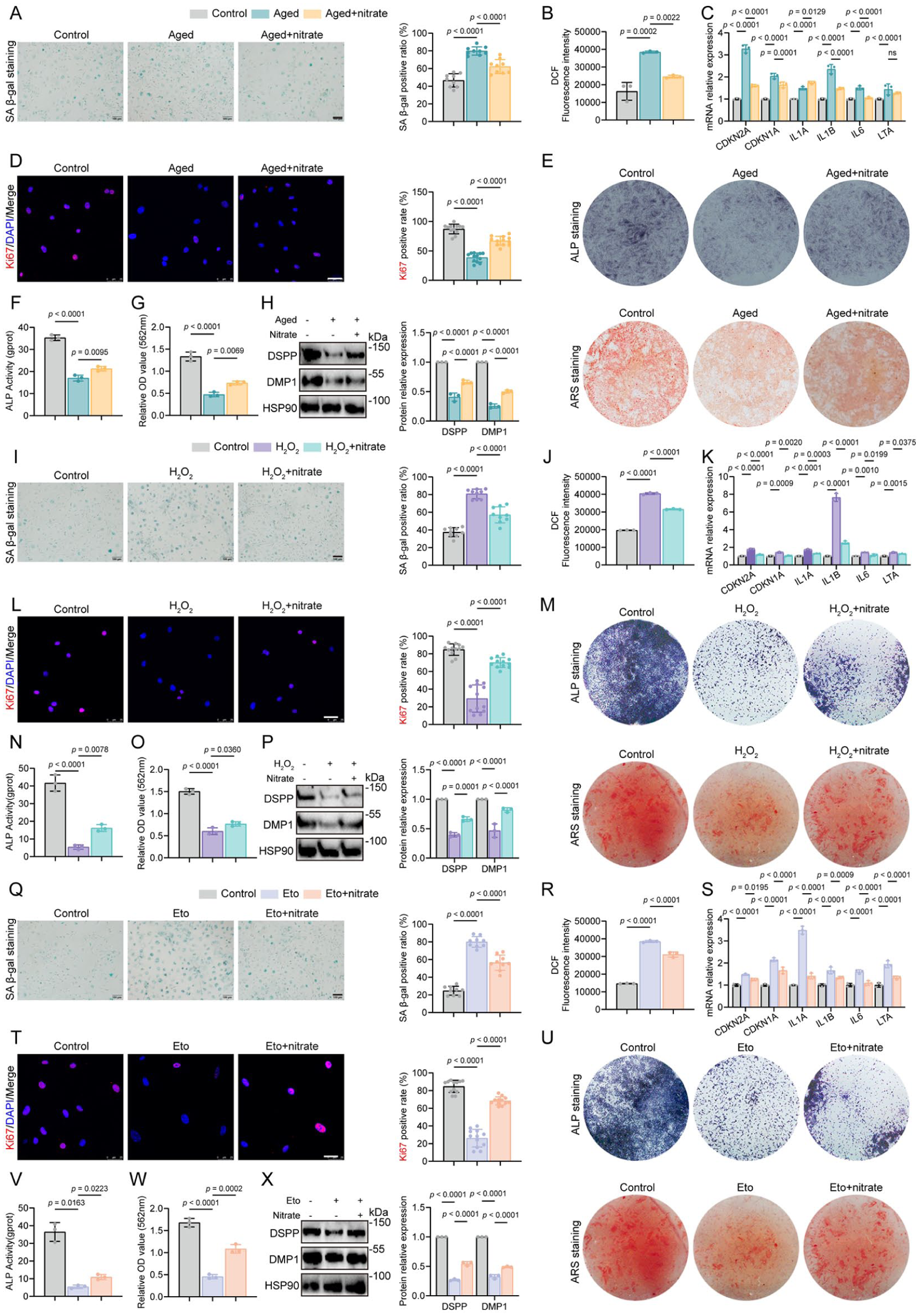
Nitrate reduces senescence and restores odontogenic capacity across 3 different models of stress in dental pulp stem cells. (
Together, these data demonstrate that nitrate mitigates senescence triggered by DNA damage, oxidative stress, or replicative aging and partially restores proliferative and odontogenic programs in DPSCs toward cellular homeostasis.
Sialin Is Required for Nitrate-Induced Antisenescence and Odontogenic Differentiation
We next tested whether the nitrate transporter Sialin (SLC17A5) is required for the observed effects. SLC17A5 knockdown accelerated senescence in DPSCs, evidenced by more SA-β-gal–positive cells, higher intracellular ROS, and fewer Ki67-positive cells (Fig. 2A–E), together with upregulation of senescence-associated genes (including CDKN2A, CDKN1A, and representative SASP genes such as IL1A, IL1B, IL6, and LTA) and proteins (Fig. 2F, N, O). In control cells, nitrate increased proliferation, reduced senescence readouts, and restored function; however, these benefits were abolished in shSLC17A5 DPSCs (Fig. 2A–F, N, O).
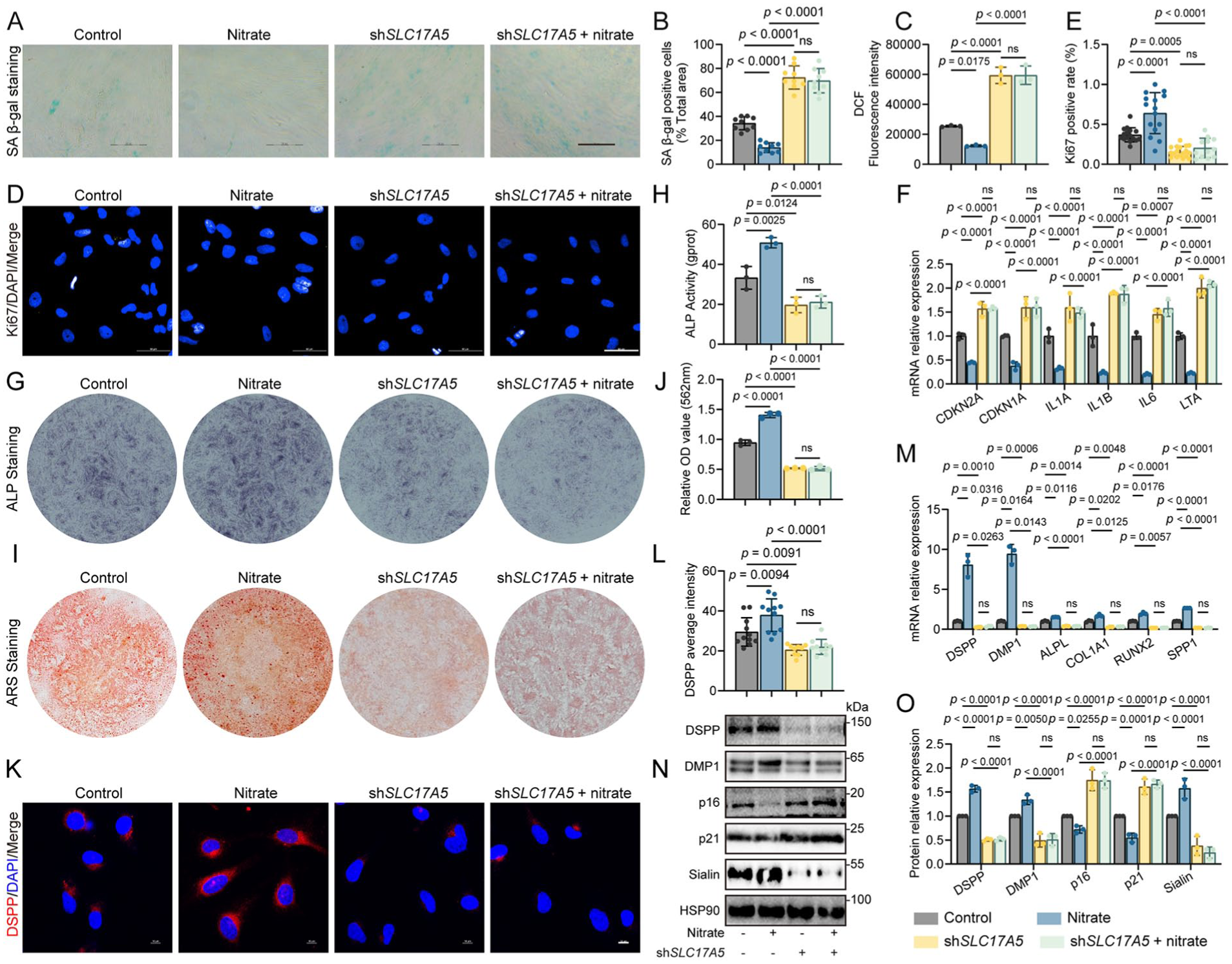
Sialin is required for nitrate-induced antisenescence and odontogenic differentiation in dental pulp stem cells. (
To determine the impact on odontogenic capacity, we quantified ALP activity and ARS mineral deposition. SLC17A5 knockdown impaired both readouts, and nitrate failed to rescue these defects under knockdown conditions (Fig. 2G–J). Consistently, odontogenic markers—DSPP, DMP1, ALPL, COL1A1, RUNX2, and SPP1—were decreased at the messenger RNA (mRNA) and protein levels after SLC17A5 knockdown, and nitrate no longer upregulated their expression (Fig. 2K–O).
Together, these data demonstrate that Sialin is necessary for nitrate-mediated antisenescence and for restoration of odontogenic differentiation and DPSC homeostasis.
Nitrate–Sialin Reprograms Mitochondrial Metabolism to Sustain DPSC Function
To define how nitrate mediates its antisenescence effects, we performed transcriptomic and metabolomic profiling. RNA sequencing (RNA-seq) revealed clear separation between control and nitrate-treated DPSCs by principal component analysis (PCA) (Appendix Fig. 4A), and Gene Ontology (GO) enrichment analysis highlighted biological processes linked to senescence control and transcriptional regulation (Fig. 3A) (Chen S et al 2025; Liu C et al 2025; Zhang et al 2025). Guided by these signals, we conducted untargeted metabolomics across conditions with or without nitrate and with SLC17A5 knockdown (Fig. 3B) (Cui et al 2025; Xiang et al 2025). PCA and correlation heatmaps showed robust group separation (Fig. 3C, E; Appendix Fig. 4B, C). Pathway and metabolite-set enrichment analyses identified pyruvate metabolism and the tricarboxylic acid (TCA) cycle as among the most significantly altered pathways, with coordinated changes in key intermediates—including citrate, α-ketoglutarate, and malate (Fig. 3D, F). These data pointed to mitochondrial metabolic rewiring as a central response to nitrate.
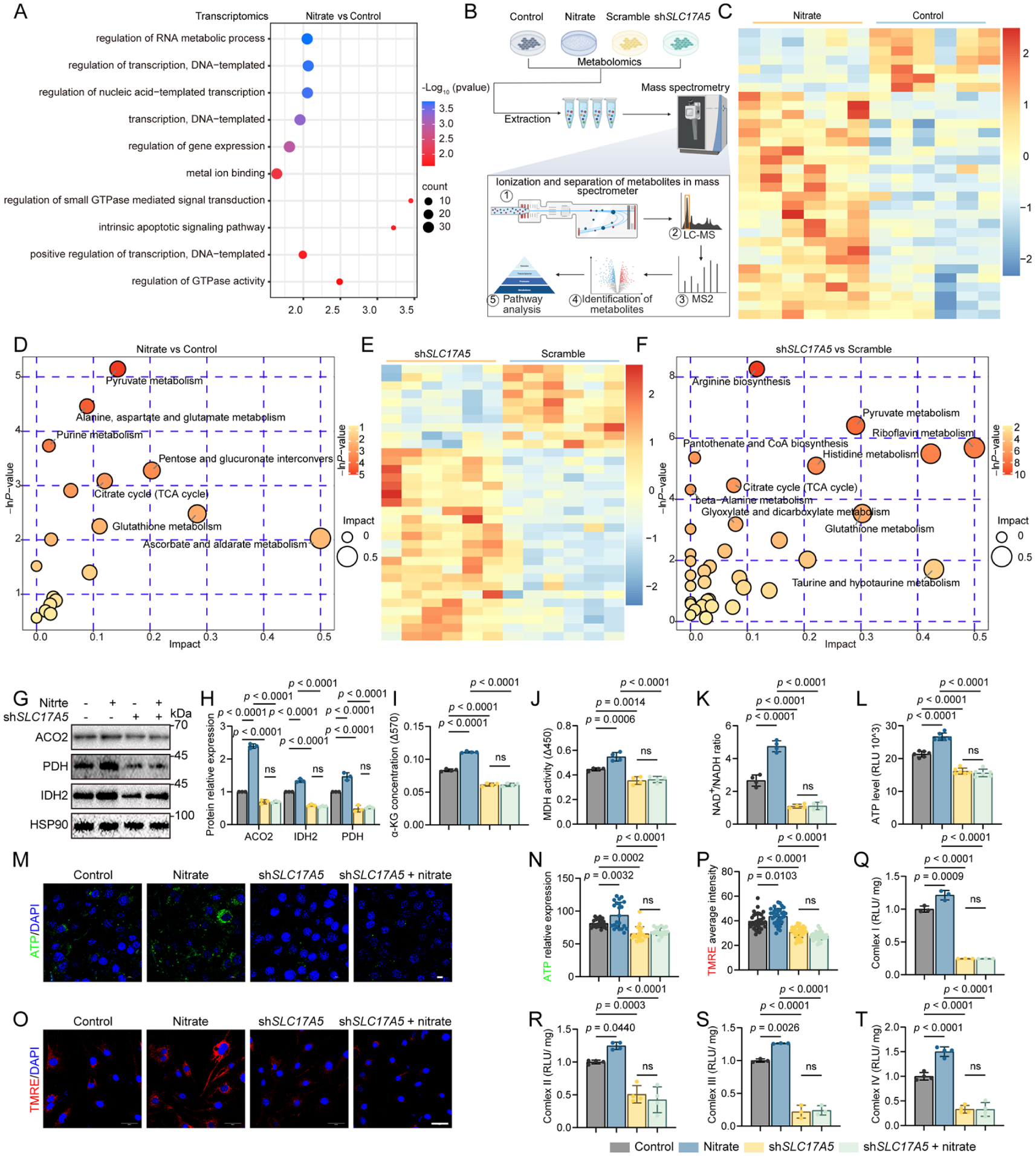
Nitrate–Sialin reprograms mitochondrial metabolism to sustain dental pulp stem cell function. (
We next validated mitochondrial function predicted by the metabolomics. Immunoblotting showed higher abundance of aconitase 2 (ACO2; a tricarboxylic acid cycle enzyme), pyruvate dehydrogenase E1α (PDH E1; linking glycolysis to the TCA cycle), and isocitrate dehydrogenase 2 (IDH2; a mitochondrial TCA enzyme) after nitrate exposure, whereas SLC17A5 knockdown reduced these enzymes and prevented their nitrate-induced increases (Fig. 3G, H). Consistent with enhanced TCA flux, α-ketoglutarate (α-KG) and malate dehydrogenase (MDH) activity were elevated, the nicotinamide adenine dinucleotide redox ratio (NAD+/NADH) increased, and both cellular adenosine triphosphate (ATP) content and mitochondrial membrane potential rose with nitrate; all changes were blunted under SLC17A5 knockdown (Fig. 3I–P). Enzymatic activities of respiratory complexes I–IV were also higher with nitrate (Fig. 3Q–T). Importantly, SLC17A5 knockdown diminished respiratory chain performance and abolished nitrate’s improvements across these mitochondrial metrics (Fig. 3Q–T).
Collectively, these results demonstrate that Sialin is necessary for nitrate-driven mitochondrial metabolism reprogramming in DPSCs, characterized by increased TCA-linked enzyme abundance, higher NAD+/NADH and ATP content, and improved respiratory complex function—hallmarks of metabolic and redox homeostasis that underpin the phenotypic rescue.
Mitochondrial Integrity Is Required for Nitrate–Sialin Effects and Is Amenable to Pharmacologic Rescue
Having established mitochondrial metabolism rewiring downstream of the nitrate–Sialin axis, we next tested whether mitochondrial function is required for nitrate to suppress senescence features and restore odontogenic function in dental pulp stem cells. We used the complex I inhibitor rotenone to impose a defined respiratory blockade and, in a complementary direction, applied GW7647 as a metabolic activator under SLC17A5 knockdown. To enable mechanistic interpretation, we first selected a rotenone concentration and exposure window that robustly inhibited complex I–linked bioenergetic readouts while avoiding overt cell collapse, as supported by viability-related control measurements shown in Appendix Fig. 5A–C (Qin et al 2022; Saller et al 2025).
Rotenone alone reduced mitochondrial function, and it prevented nitrate-driven metabolic and phenotypic improvements. Immunoblotting showed that nitrate-induced increases in ACO2, PDH E1, and IDH2 were not observed when rotenone was present (Fig. 4A, B). Consistently, α-KG concentration, MDH activity, NAD+/NADH ratio, and ATP production remained suppressed under rotenone exposure irrespective of nitrate treatment (Fig. 4C–F). Activities of mitochondrial respiratory chain complexes I–IV were reduced (Fig. 4G–J), and mitochondrial superoxide measured by MitoSOX remained elevated under rotenone conditions (Fig. 4K, L).
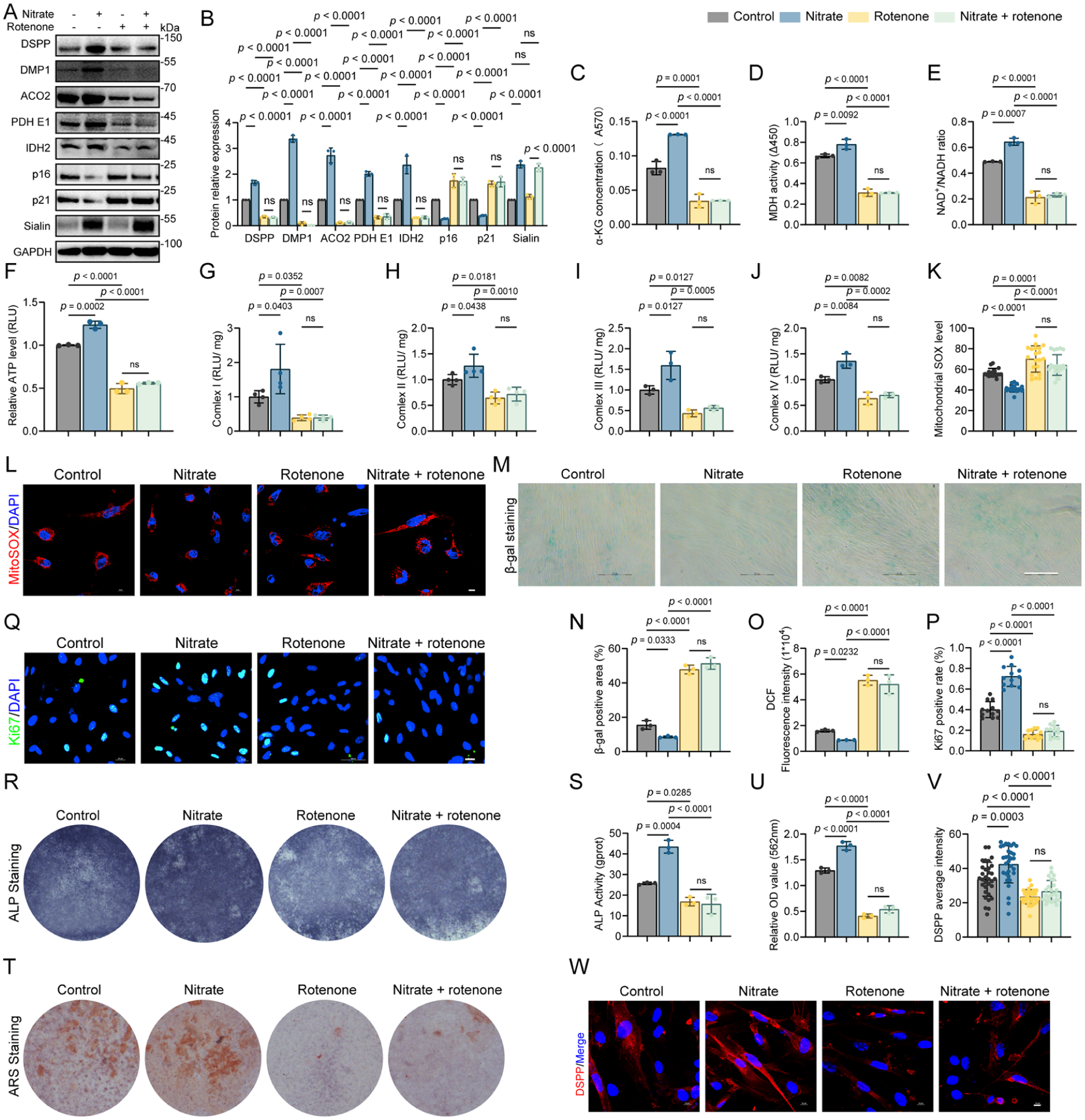
Mitochondrial inhibition by rotenone abolishes nitrate-driven antisenescence and odontogenic responses in dental pulp stem cells. (
At the phenotypic level, rotenone maintained senescence-associated outputs and blocked nitrate-dependent rescue. SA-β-gal positivity remained high; senescence-associated genes, including CDKN2A, CDKN1A, and SASP transcripts such as IL1A, IL1B, IL6, and LTA, remained elevated; and p16 and p21 protein abundance increased, accompanied by a reduced fraction of Ki67-positive cells (Fig. 4A, M–Q; Appendix Fig. 5D). Odontogenic endpoints, including ALP activity, ARS mineral deposition, and expression of odontogenic markers, were similarly diminished under rotenone exposure, and nitrate did not restore these outcomes (Fig. 4A, R–W; Appendix Fig. 5E). Across the principal mitochondrial and differentiation readouts, rotenone-only and nitrate with rotenone groups were not statistically different, indicating that nitrate’s beneficial effects require functional electron transport under the conditions tested.
Metabolic activation partially rescues deficits in SLC17A5 knockdown cells. In SLC17A5 knockdown DPSCs, GW7647 increased the abundance of ACO2, PDH E1, and IDH2 (Appendix Fig. 6A) and improved multiple TCA-linked and energetic indices, including higher α-KG and improved NAD+/NADH and ATP production (Appendix Fig. 6B–E). Consistently, activities of mitochondrial respiratory complexes I–IV were restored toward control levels (Appendix Fig. 6F–I). These mitochondrial improvements were accompanied by a functional recovery at the cellular level, including reduced senescence-associated readouts (Appendix Fig. 6J–O), increased proliferative activity (Appendix Fig. 6P, Q), and improved odontogenic differentiation outcomes measured by mineralization assays and differentiation marker expression (Appendix Fig. 6A, R–W). These effects indicate that augmenting mitochondrial function can offset, at least in part, the deficits caused by loss of Sialin.
Together, these data indicate that enhancing mitochondrial metabolic capacity can compensate, at least in part, for the loss of SLC17A5 and support mitochondrial function as an actionable node linking the nitrate–Sialin axis to preservation of DPSC functional homeostasis.
Mesenchymal Lineage–Specific Sialin Deficiency Abrogates Nitrate-Induced Dentin Regeneration In Vivo
To determine whether the reparative effect of nitrate in vivo requires Sialin in the mesenchymal lineage, we analyzed Prx1–Cre; Slc17a5fl/fl conditional knockout (cKO) mice alongside wild-type controls and verified efficient loss of Sialin signal in the dental pulp stromal compartment in uninjured molars (Appendix Fig. 7A, B). Micro–computed tomography demonstrated that wild-type (WT) mice receiving nitrate formed a continuous dentin bridge–like hard tissue at the exposure site, whereas WT mice without nitrate showed only limited peripheral mineral deposition, and cKO mice—regardless of nitrate exposure—exhibited minimal hard tissue formation (Fig. 5A). Quantitative analysis confirmed superior mineral repair in the WT + nitrate group, reflected by higher bridge mineral density (BMD) and mineralized bone volume/total volume (BV/TV) than WT without nitrate, whereas both cKO groups, irrespective of nitrate, remained near baseline and exhibited greater dentin loss than WT controls (Fig. 5B, C).
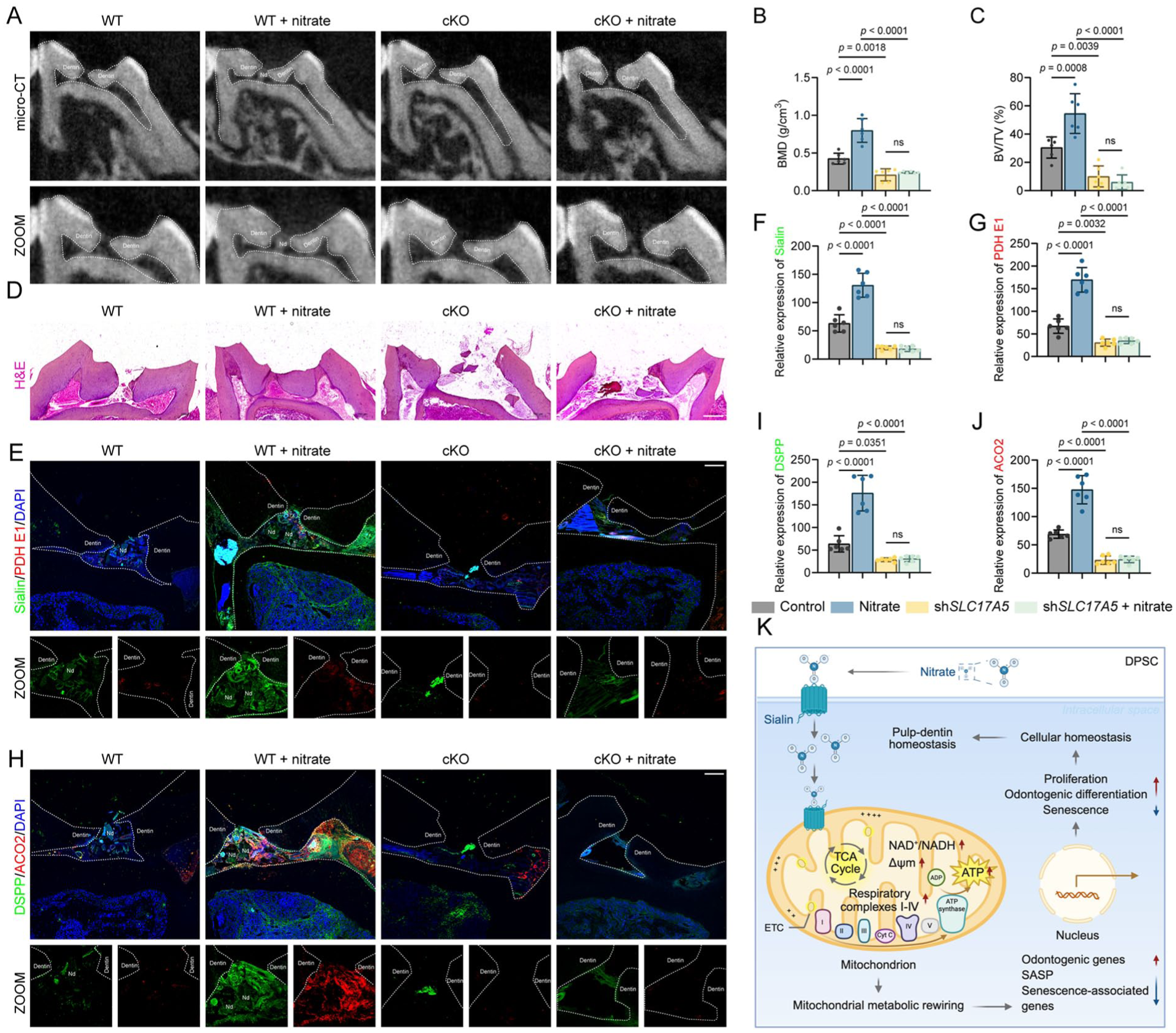
Mesenchymal lineage–specific Sialin deficiency abrogates nitrate-induced dentin regeneration in vivo. (
Histological findings corroborated the imaging results. Hematoxylin and eosin (H&E) staining revealed sparse or no mineralized tissue in WT mice without nitrate, with only discontinuous deposits along the exposure margins (Fig. 5D). In contrast, the WT + nitrate group exhibited a well-organized reparative dentin bridge that sealed the exposure site, characterized by continuous mineralized tissue with aligned dentinal tubules (Fig. 5D). Both cKO groups—irrespective of nitrate—exhibited minimal or absent mineral deposition, and the exposure sites remained largely unhealed (Fig. 5D). Furthermore, the WT + nitrate group showed strong signals for Sialin, DSPP, PDH E1, and ACO2 (Fig. 5E–J), whereas WT without nitrate displayed weaker staining, and both cKO groups showed markedly reduced or undetectable expression of these proteins.
Together, these findings demonstrate that nitrate promotes reparative dentin formation in vivo in a Sialin-dependent manner, supporting recovery of pulp–dentin homeostasis (Fig. 5K).
Discussion
Our study tested whether nitrate could counteract cellular senescence and restore DPSC function, and we demonstrate that nitrate supplementation consistently attenuates senescence and reinstates odontogenic capacity across 3 complementary models. Loss of function of Sialin (SLC17A5) establishes pathway necessity, and multilayer metabolic analyses support a mechanism involving mitochondrial rewiring rather than solely changes in the extracellular milieu (Shannon et al 2022; Chen X et al 2025). Together, these data define a nitrate–Sialin–mitochondria axis that stabilizes DPSC homeostasis and regenerative competence.
A pivotal implication of this work is the reframing of nitrate from a passive dietary substrate to an upstream signal that modulates stem cell fate via Sialin. Mitochondrial bioenergetics is a well-established determinant of stem cell fate and senescence. Whereas prior studies emphasized vascular or nitric oxide–centered effects of nitrate, our findings identify a Sialin-dependent route that specifically converges on mitochondrial function in the dental pulp context (Zhu et al 2021). This is conceptually significant because Sialin, classically annotated as a vesicular anion transporter, here emerges as a gatekeeper for a pro-regenerative metabolic program (Hu et al 2023; Schmiege et al 2024). By integrating untargeted metabolomics with mitochondrial readouts, we observe increased TCA-linked flux, elevated ATP, a higher NAD+/NADH ratio, lower mitochondrial superoxide, and improved respiratory complex performance, consistent with nitrate acting as an upstream, transporter-defined lever on mitochondrial metabolism in senescent DPSCs. Importantly, our pharmacologic perturbation data support a perturbation–response framework in which mitochondrial electron transport is required for the nitrate phenotype, rather than a nonspecific secondary consequence of increased proliferation. These results extend current strategies that target mitochondria in aging tissues by leveraging a biocompatible, low-cost, and locally deliverable small anion.
The biological and translational impacts are 2-fold. First, at the cell-intrinsic level, Sialin-mediated nitrate handling provides a tractable lever to correct the energetic and redox defects that lock senescent DPSCs into low-odonto states. Second, at the tissue level, local administration offers a practical means to achieve effective exposure while minimizing systemic load. Given the donor-to-donor heterogeneity that limits stem cell–based interventions, a transporter-anchored metabolic approach may help normalize functional outputs and reestablish tissue homeostasis across variable starting states, suggesting broader applicability to stem/progenitor compartments beyond the pulp–dentin complex. In this framework, nitrate operates not as a generic antioxidant but as a cue that reestablishes mitochondrial efficiency within physiologic ranges compatible with differentiation.
Despite the mechanistic and in vivo evidence presented, several limitations should be clarified. Our study was designed to resolve a Sialin-dependent nitrate program that restores odontogenic competence and supports reparative dentin formation; accordingly, we did not extend the study to cross-lineage differentiation assays, and whether nitrate–Sialin signaling influences adipogenic or chondrogenic potential remains an important question for future work. In the in vivo pulp exposure setting, the evidence centers on pathway engagement and outcome—mitochondrial metabolic marker induction together with dentin bridge formation and odontogenic markers—while direct pharmacodynamic measurements, including stromal cell nitrate uptake and broader nitrate-responsive gene signatures in oral tissues, were not performed and will be valuable to establish in situ target engagement. Likewise, canonical senescence markers such as p16, p21, or γH2AX were not quantified in the pulp sections available from this long-term experiment, and future studies should incorporate dedicated sampling to link senescence resolution more directly to tissue repair. Finally, although our model used a capping seal to maintain the injury environment, we did not benchmark nitrate delivery against clinically established calcium–silicate materials, such as mineral trioxide aggregate or related cements; head-to-head comparisons will be essential to contextualize efficacy and to define whether nitrate is best positioned as a stand-alone local cue or as an adjunct that enhances cellular fitness within contemporary vital pulp therapy workflows.
In summary, we delineate a nitrate–Sialin–mitochondria axis that mitigates DPSC senescence and restores odontogenic function to rebuild pulp–dentin homeostasis. By coupling a readily deployable inorganic cue to organelle-level control of stem cell fate, this work provides both a mechanistic rationale and a translational path for vital pulp therapy. Clinically, these findings suggest an actionable strategy to improve the quality and predictability of reparative dentin formation by targeting the pulp stromal and progenitor compartment, a key determinant of durable treatment success. Because nitrate is biocompatible, inexpensive, and amenable to local administration, it may be developed as an adjunct to contemporary pulp-capping materials or regenerative endodontic approaches to enhance cellular fitness in inflamed or aging-prone pulps and thereby support more consistent dentin bridge formation. Future studies optimizing formulation, dose–time profiles, and delivery vehicles in clinically relevant injury settings will be important for translation.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261435516 – Supplemental material for Nitrate–Sialin Promotes Dentin Regeneration via Mitochondrial Rewiring
Supplemental material, sj-docx-1-jdr-10.1177_00220345261435516 for Nitrate–Sialin Promotes Dentin Regeneration via Mitochondrial Rewiring by Z. Cao, S. Kong, O. Jiang, J. Wang, S. Wang and X. Li in Journal of Dental Research
Footnotes
Acknowledgements
We thank the Core Facility Center, Capital Medical University, and the Laboratory for Clinical Medicine, Capital Medical University, for technical support. Scheme is generated using BioRender.
Author Contributions
Z. Cao, contributed to conception and design, data analysis and interpretation, drafted and critically revised the manuscript; S. Kong, contributed to conception and design, data acquisition and interpretation, drafted and critically revised the manuscript; O. Jiang, contributed to data analysis, drafted manuscript; J. Wang, S. Wang, contributed to data interpretation, critically revised the manuscript; X. Li, contributed to conception and design, data analysis, critically revised the manuscript. All authors gave their final approval and agreed to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: X.L. is supported by grants from the National Natural Science Foundation of China (82401082), the State Key Laboratory of Oral Diseases Open Fund (SKLOD2023OF11), Young Scientist Program of Beijing Stomatological Hospital, Capital Medical University (YSP202308), and Beijing High-Level Innovation and Entrepreneurship Talent Support Program—Dengfeng Project (G202512061). S.W. is supported by grants from the Beijing Municipal Government grant (Beijing Laboratory of Oral Health, PXM2021_014226_000041 and PXM2021_014226_000020; Beijing Scholar Program-PXM2018_014226_000021), the National Natural Science Foundation of China (82030031 and 92149301), and Chinese Research Unit of Tooth Development and Regeneration, Academy of Medical Sciences (No. 2019-12M-5-031).
Data Availability
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.