
Abstract
Oral leukoplakia (OLK), the most common potentially malignant oral disorder, poses a persistent clinical challenge due to high recurrence and malignant transformation rates, with no curative treatment currently available. Conventional interventions, including medications, surgery, laser, and photodynamic therapy, have shown limited success in cancerization prevention, largely because the abnormal events underlying the development and progression of OLK remain poorly defined. Here, through single-cell RNA sequencing profiles, we identified an OLK-specific cell population characterized by YAP/TAZ overactivation, which was strongly associated with disrupted epithelial homeostasis. Transgenic mouse models confirmed the essential role of YAP/TAZ activation in conferring the malignant phenotype of OLK and driving its progression toward transformation. Interestingly, although YAP/TAZ are technically and strategically considered undruggable, we uncovered a ferroptosis vulnerability in OLK malignant cells mediated by the YAP/TAZ-TEAD-TXNRD1 axis. Notably, we demonstrate that combining sulfasalazine and docosahexaenoic acid, 2 clinically available agents, effectively induces ferroptosis and attenuates OLK progression. These findings establish a mechanistic link between YAP/TAZ activation and OLK progression, specifically revealing the dual roles of YAP/TAZ in maintaining malignant properties while conferring ferroptosis vulnerability. This duality highlights ferroptosis induction as a promising therapeutic strategy for indirectly targeting YAP/TAZ. Moreover, the drug-repurposing approach provides a practical and translationally relevant avenue to improve OLK management and reduce the risk of malignant transformation.
Keywords
Introduction
Oral leukoplakia (OLK) presents as a white lesion occurring on the surface of oral mucosa and represents the most common potentially malignant oral disorder. When compared with the general population, patients with OLK have a 40.8-times higher risk of developing oral squamous cell carcinoma (Chaturvedi et al 2020). Current treatment strategies for OLK can be broadly classified into surgical interventions and pharmacologic approaches. Surgical excision, laser therapy, cryotherapy, and photodynamic therapy are effective in removing visible lesions but are associated with unpredictable risks of recurrence and malignant transformation (MT; Sundberg et al 2019; Paglioni et al 2020; Jing et al 2024). This challenge largely stems from the presence of molecularly abnormal cells beyond visibly affected areas, complicating margin delineation and therapeutic precision. Topical and systemic medications , such as retinoid, beta-carotene, and vitamins, frustratingly demonstrate no evidence of effectiveness in preventing MT and even have a higher recurrence rate than surgical interventions (Ribeiro et al 2010). A fundamental limitation of current pharmacological therapies is the lack of a clearly defined mechanistic understanding of functional contributions during the initiation and progression of OLK. As a result, these treatments remain largely symptom oriented, primarily aimed at alleviating clinical manifestations rather than targeting the underlying disease drivers.
Epithelial architectural disruption is a pathologic feature of OLK, encompassing hyperplasia and keratosis or epithelial dysplasia. The presence of epithelial dysplasia is an important indicator of the malignant potential of OLK, and clinical evidence reveals that the risk of progression to oral squamous cell carcinoma increases with higher dysplasia grades. Although prior research using bulk tissue analyses has revealed distinct molecular profiles that differentiate dysplastic from nondysplastic OLKs (Farah and Fox 2019), it has been unable to identify the specific cell populations within OLK lesions that undergo MT or to elucidate the underlying dynamic biological processes.
Here, we identified that the expansion of basal-like epithelial cell populations is a defining feature of OLK. YAP/TAZ overactivation enables epithelial cells to hyperproliferate and promote malignant progression. Rather than directly targeting YAP/TAZ signaling, we leveraged the ferroptosis vulnerability created by YAP/TAZ overactivation in these basal-like epithelial cells, highlighting ferroptosis induction as a promising therapeutic strategy for OLK treatment and MT prevention.
Materials and Methods
This study employed single-cell RNA sequencing (RNA-seq) analysis to characterize aberrant molecular events in OLK and generated conditional transgenic mouse models to elucidate the role of YAP/TAZ-TEAD (transcriptional enhanced associate domain) signaling in OLK progression. Bioinformatic analysis integrated with molecular analyses identified potential therapeutic targets linked to aberrant YAP/TAZ activation. Finally, a drug repurposing strategy was applied to evaluate the therapeutic efficacy and translational potential of ferroptosis-inducing treatment for OLK in in vitro and in vivo models. Detailed materials and methods are provided in the Appendix.
Results
Basal-like Populations Expanded in OLK
To unveil the characterization of abnormal cell populations in OLK, we interrogated a publicly available single-cell RNA-seq (scRNA-seq) dataset (Choi et al 2023), comparing transcriptional profiles of dysplastic OLK and normal oral mucosa. Uniform manifold approximation and projection separated by classical cell markers revealed 9 distinct clusters, primarily comprising immune cells, fibroblasts, epithelial cells, and endothelial cells (Fig. 1A). For a typical epithelial lesion, we focused on the epithelial cell populations, where we observed notable differences between OLK and normal tissues (Fig. 1B). Clusters 0 and 9 consist primarily of cells from OLK samples, whereas the remaining clusters mainly belong to normal mucosa (Fig. 1B, Appendix Fig. 1A). This distribution pattern suggests that OLK possesses unique transcriptomic features distinguishing it from normal epithelium. Differential expression gene (DEG) analysis, comparing clusters 0 and 9 against all other clusters, identified keratin 14 (KRT14) and KRT5 as the most highly upregulated genes (Fig. 1C, Appendix Fig. 1B). KRTs are the largest subgroup of intermediate filament proteins, preferentially expressed in epithelial tissues (Alam et al 2011). KRT14 and KRT5 are coexpressed in mitotically active basal cells of stratified epithelia, which contain stem-like and transient amplifying cells that maintain epithelial homeostasis. As these cells migrate upward and differentiate, KRT14/KRT5 expression gradually decreases. However, a substantial expansion of cell populations coexpressing KRT5/KRT14 was observed in OLKs (Fig. 1D), and immunohistochemical staining consistently revealed that increased KRT14 expression extended into the suprabasal layers, indicating disrupted epithelial architecture and aberrant differentiation (Fig. 1E, Appendix Fig. 1C). Together, these findings demonstrate disrupted epithelial homeostasis in OLKs, characterized by an expansion of basal-like cell populations.
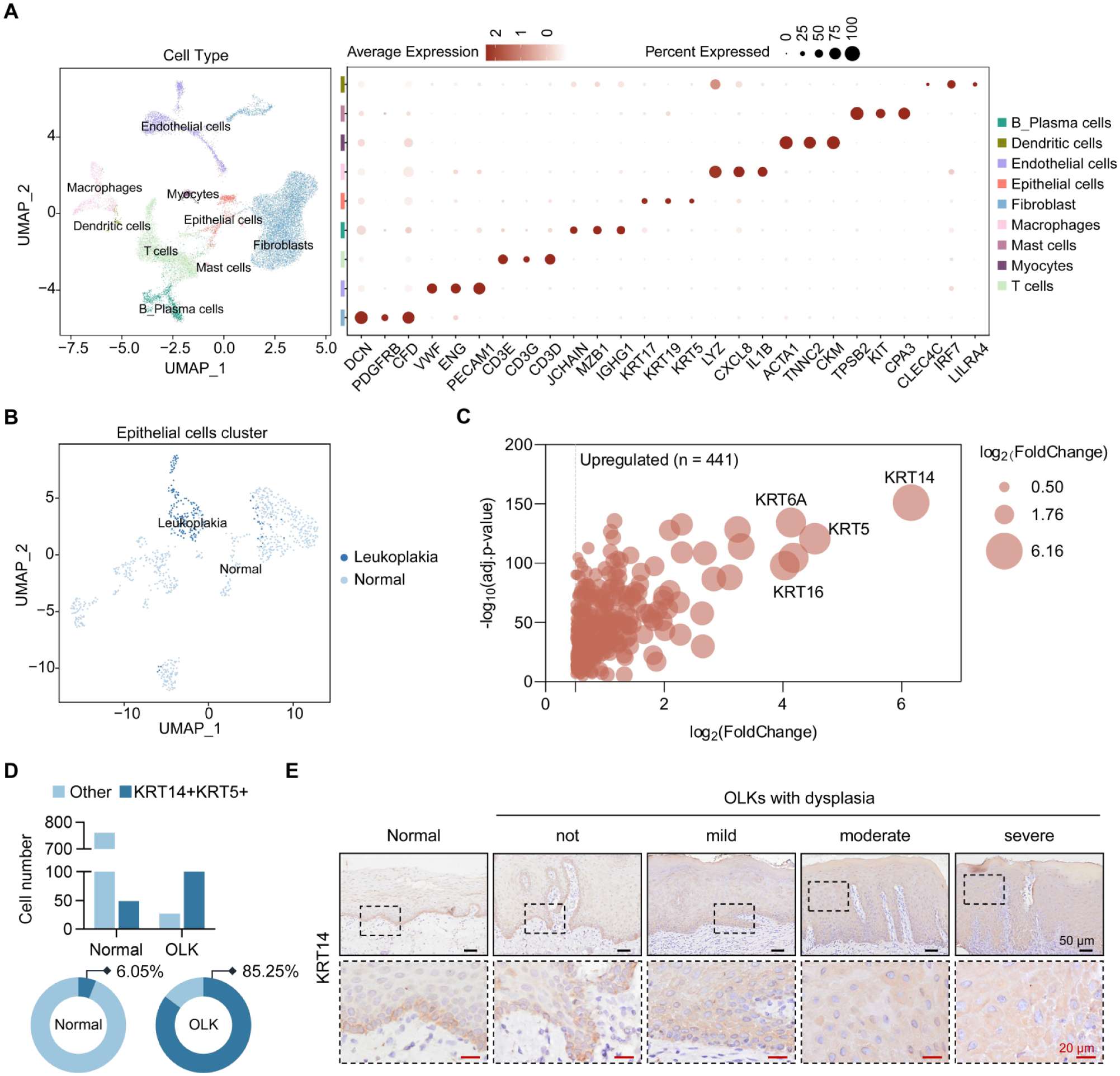
Single-cell RNA sequencing analysis reveals disruption of epithelial homeostasis in oral leukoplakia (OLK). (
YAP/TAZ Overactivation Underlies OLK Epithelial Disruption
To investigate the molecular mechanisms underlying disrupted epithelial homeostasis in OLK, we analyzed publicly available bulk RNA-seq data containing 22 OLK samples with dysplasia and 18 normal samples (Khan et al 2023). Similar to the scRNA-seq findings, DEG analysis revealed significant upregulation of KRT14, KRT16, and KRT75 in OLK (Fig. 2A). In contrast, KRT4, a well-established marker of epithelial differentiation, was the most significantly downregulated gene (Fig. 2A), reflecting impaired differentiation and enhanced basal-like KRT expression in OLK. Given that transcription factors often act as pivotal regulators in specific biological processes, we performed transcription factor prediction on the identified DEGs, where TEAD2 ranked highest, with TEAD4 also appearing within the top 10 candidates (Fig. 2B). As canonical downstream effectors of YAP/TAZ signaling, the enrichment of TEADs (ie, transcriptional enhanced associate domains) suggests the possible involvement of YAP/TAZ in driving the transcriptional reprogramming underlying OLK. Supporting this, we found several canonical YAP/TAZ target genes also upregulated in OLK samples, including CCN1, CCN2, and SERPINE1 (Wang et al 2018; Fig. 2A, Appendix Fig. 2A), as well as an enrichment of YAP-conserved signature based on gene set enrichment analysis (Fig. 2C). YAP was particularly and consistently upregulated in OLK-enriched populations based on scRNA-seq (Appendix Fig. 1B). Histologically, YAP/TAZ was activated but confined to the basal layer in normal mucosa; in contrast, OLK lesions demonstrated significantly elevated expression that breached this spatial restriction, and this overexpression progressively intensified from mild to severe dysplasia (Fig. 2D, Appendix Fig. 2B).
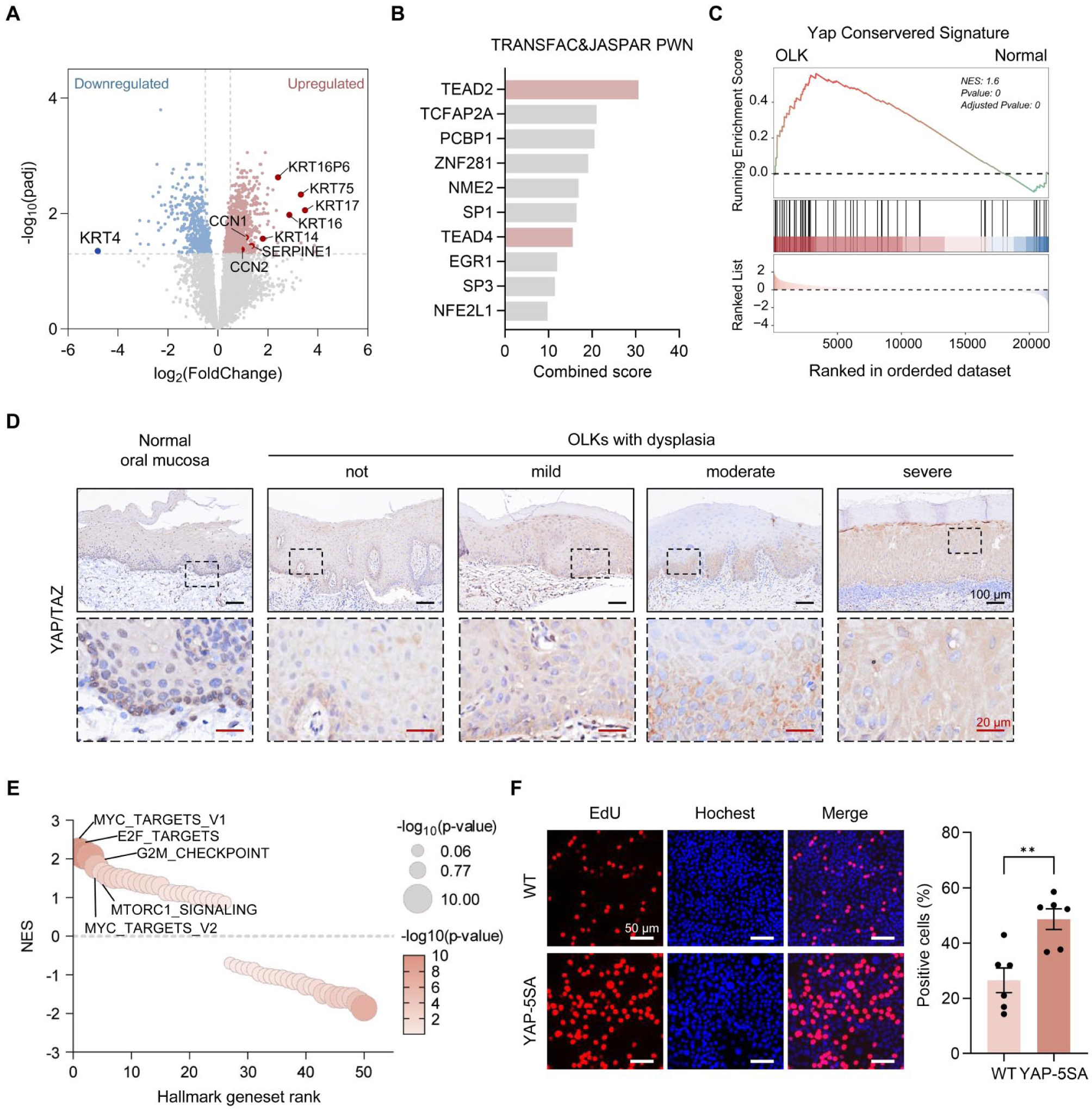
YAP is overactivated in oral leukoplakia (OLK) and able to drive hyperproliferation. (
YAP/TAZ are critical regulators of diverse biological processes, including cell proliferation, development, stem cell maintenance, and tumorigenesis (Zanconato et al 2016). Previous studies have established their function with transcriptional factor TEADs in maintaining the basal cell state, while TEAD inhibition promotes cell differentiation (Yuan et al 2020). Interestingly, gene set enrichment analysis of the hallmark gene sets revealed significant enrichment of the MYC targets, E2F targets, and G2M checkpoint pathways in OLK samples (Fig. 2E), indicating enhanced cell cycle progression and proliferative signaling. YAP is inactivated by phosphorylation at multiple serine residues mediated by Hippo signaling. To investigate the effects of sustained YAP activation in epithelial cells, we generated cells expressing a constitutively active YAP mutant (YAP-5SA), in which 5 serine residues were substituted with alanine to prevent phosphorylation and promote persistent transcriptional activity. Elevated expression of CCN1 (CYR61) and CCN2 (CTGF) confirmed enhanced activity in YAP-5SA cells (Appendix Fig. 2C, D). Notably, YAP-5SA cells maintained high proliferative capacity despite touch inhibition, a condition normally suppressed by Hippo signaling (Zhao et al 2007; Fig. 2F). Collectively, these findings indicate that YAP/TAZ are overactivated in OLKs, prompting basal-like cell hyperproliferation.
Inhibition of YAP/TAZ-TEAD Signaling Attenuates the Malignant Progression of OLK
Next, we questioned whether YAP/TAZ overactivation contributes to OLK progression. Given the expansion of KRT14+ cells beyond the basal layer, we employed the KRT14 promoter to drive tamoxifen-inducible Cre-recombinase (KRT14CreERT) expression in transgenic mice. SuperHippo is an engineered minigene that activates LATS1/2, effectively mimicking the complete loss function of YAP/TAZ (Qi et al 2022). Then, we crossed a SuperHippo transgenic mouse with KRT14CreERT to generate a basal epithelium–specific conditional mouse model. Littermates bearing KRT14CreERT but wild type for SuperHippo served as controls (Appendix Fig. 3). After exposure to 4-NQO in drinking water for 16 wk to established OLK-like lesions on the tongue, tamoxifen was administered to the mice via intraperitoneal injection to induce the activation of SuperHippo (Fig. 3A). Following a 6-wk observation, OLK-like lesions in controls with preserved YAP/TAZ activity gradually protruded above the mucosal surface, with several rapidly advancing to oral squamous cell carcinoma (Fig. 3B). In contrast, lesion progression in SuperHippo mice was markedly attenuated, showing reduced dysplasia and, in some cases, complete regression (Fig. 3B, C, and F), suggesting that loss of YAP/TAZ function delays the MT of OLK.
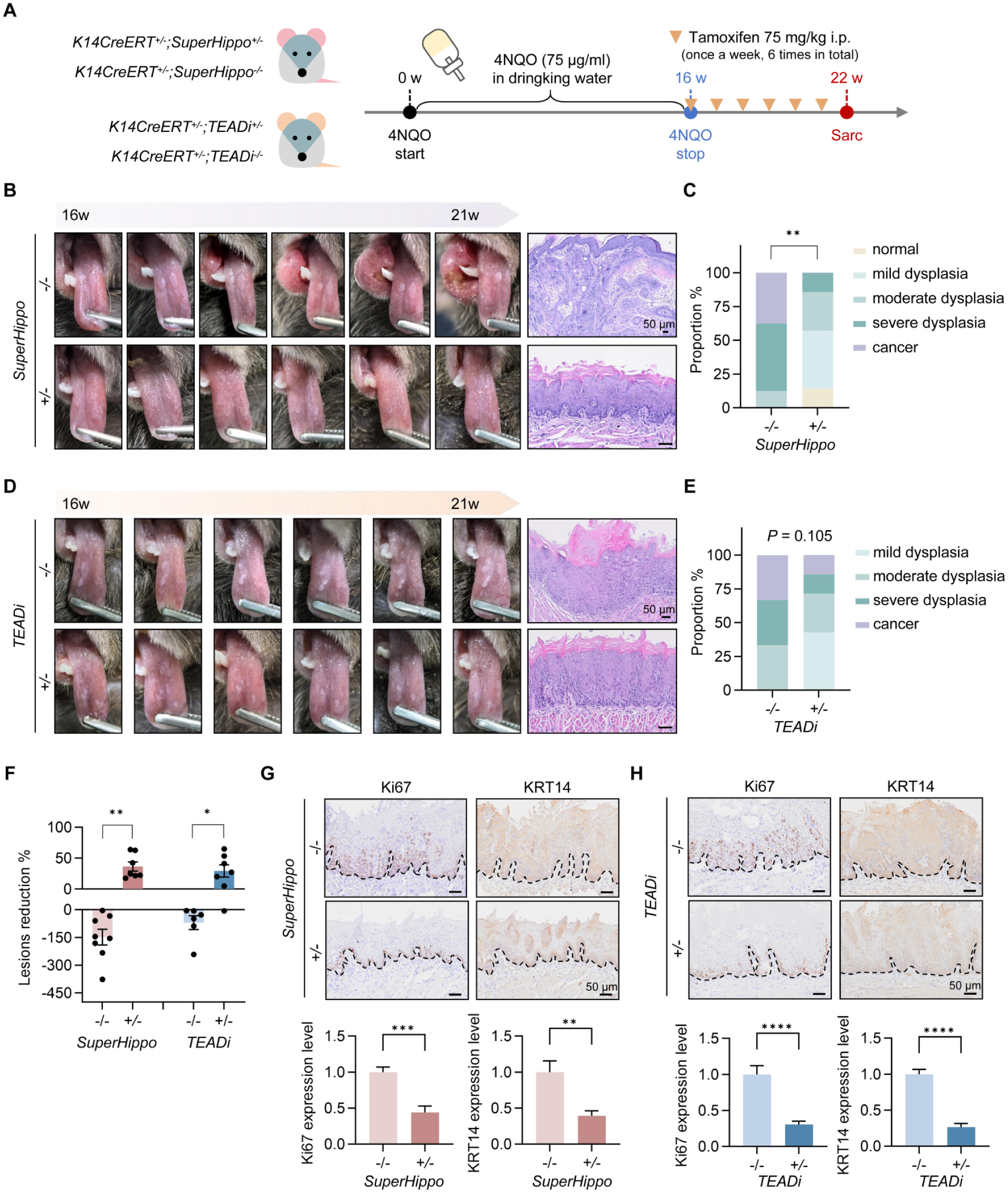
YAP/TAZ-TEAD (transcriptional enhanced associate domain)signaling drives oral leukoplakia (OLK) progression in transgenic mice. (
TEAD-dependent transcriptional networks have been implicated in epithelial state initiation (Yuan et al 2020). To determine whether YAP/TAZ-driven OLK progression is mediated through TEAD, we introduced a tamoxifen-inducible TEADi transgenic mouse model also under KRT14 control (Fig. 3A, Appendix Fig. 3). TEADi is a peptide that can block the interaction with YAP/TAZ via competitively binding to TEAD (Yuan et al 2020). Similar to the SuperHippo group, TEADi mice displayed smoother white plaques, thinner stratum corneum, and mild to moderate dysplasia as compared with the wild type group (Fig. 3D–F). The pathologic improvement was less pronounced than in SuperHippo mice, suggesting that YAP/TAZ promote OLK progression through additional TEAD-independent regulations.
Moreover, SuperHippo and TEADi expression resulted in reduced basal cell proliferation, as indicated by decreased Ki67 staining (Fig. 3G, H). In addition, KRT14 expression became largely restricted to the basal layer, suggesting partial restoration of epithelial homeostasis (Fig. 3G, H). Together, these findings suggest that YAP/TAZ-TEAD is a crucial hub controlling OLK malignant progression.
YAP/TAZ Promote Ferroptosis Sensitivity through TXNRD1 in OLK
Although YAP/TAZ-TEAD inhibition can mitigate OLK malignant progression, directly targeting them seems risky given their indispensable physiologic roles (Piccolo et al 2014; Feng 2021; Driskill and Pan 2023). Therefore, we shifted our focus to identify alternative and targetable vulnerabilities in OLK. Gene set enrichment analysis of the aforementioned scRNA-seq data was performed to find initial clues of alterations. Notably, this analysis revealed enrichment of pathways related to redox regulation and lipid metabolism in OLK-enriched clusters, such as reactive oxygen species, fatty acid metabolism, and adipogenesis (Fig. 4A, Appendix Table 3), suggesting that oxidative stress and altered lipid metabolic processes may represent additional features of OLK. Given that lipids are highly susceptible to peroxidation under excessive intracellular reactive oxygen species, we performed immunohistochemical staining for 4-hydroxynonenal (4HNE), a well-known toxic product of lipid peroxidation. When compared with normal mucosa, marked accumulation of 4HNE was found in human OLK and 4NQO-induced OLK tissues (Fig. 4B, Appendix Fig. 4A). Interestingly, lipid peroxidation and redox imbalance are central to ferroptosis (Dixon and Olzmann 2024). We therefore sought to validate whether ferroptosis-related programs were broadly activated in OLK. Using bulk RNA-seq to obtain cohort-level evidence, we indeed found enrichment of ferroptosis-associated pathways in OLK, suggesting that OLK may exhibit altered ferroptosis sensitivity (Dixon and Olzmann 2024; Appendix Fig. 4B). These bioinformatic clues are exactly consistent with our previous study, in which erastin was more effective in OLK cells than in normal oral keratinocytes (Yang et al 2024). Moreover, erastin-treated OLK cells were confirmed to undergo ferroptosis, as a ferroptosis inhibitor, not necrosis or apoptosis inhibitor, rescued erastin-induced cell death (Appendix Fig. 4C).
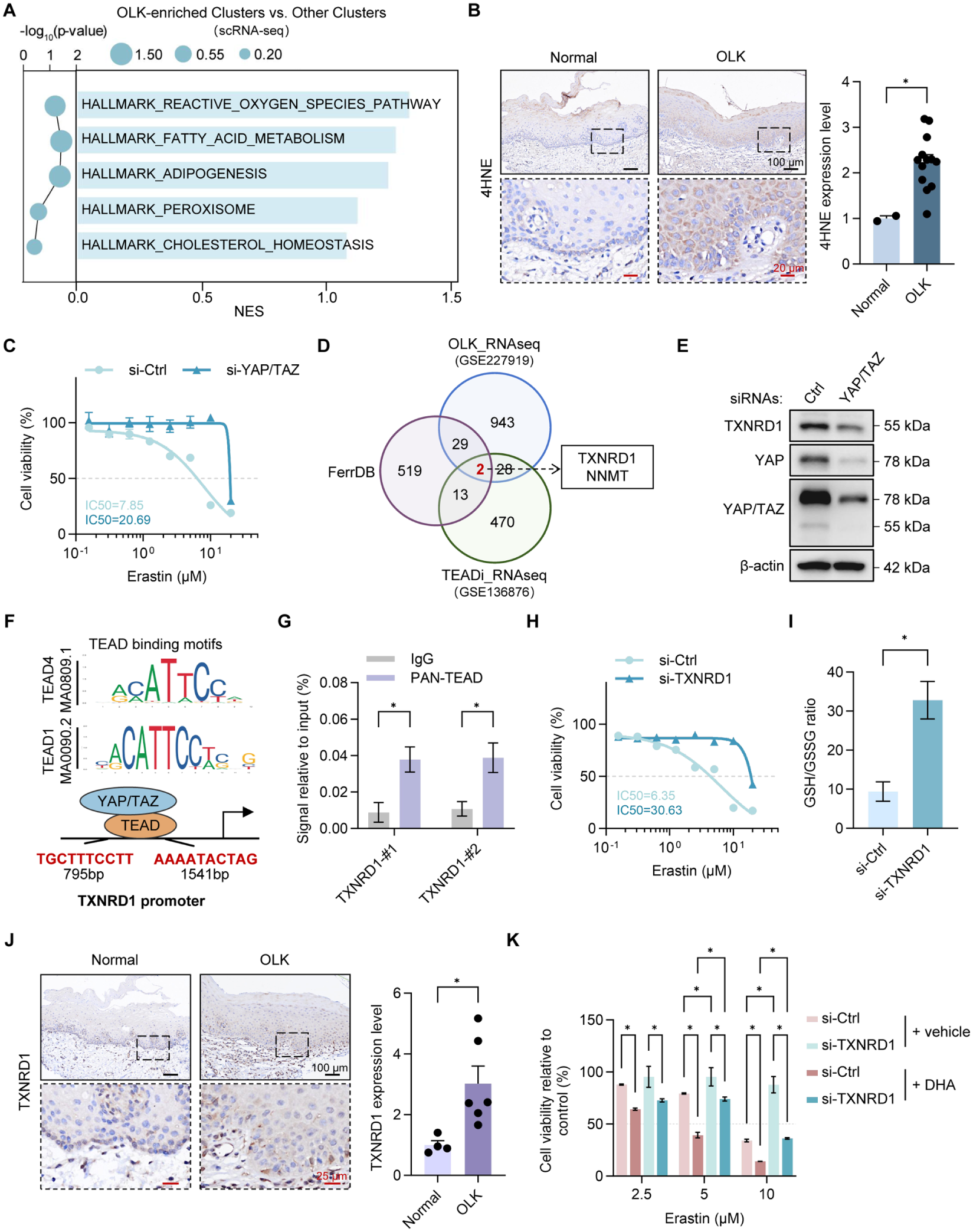
Oral leukoplakia (OLK) exhibits YAP/TAZ-dependent ferroptosis sensitivity mediated by TXNRD1. (
Since OLK-enriched clusters in scRNA-seq have a high expression level of YAP/TAZ, we next asked whether YAP/TAZ regulates this altered ferroptosis sensitivity. Surprisingly, siRNA-mediated YAP/TAZ knockdown increased the half-maximal inhibitory concentration for erastin (Fig. 4C), confirming our speculation. To elucidate their regulatory relationship, we took the intersection of OLK-upregulated DEGs from bulk RNA-seq (Khan et al 2023), DEGs downregulated by YAP/TAZ inhibition via TEADi (Yuan et al 2020), and ferroptosis-related genes from the FerrDB database (Zhou et al 2023), which yielded a promising candidate: thioredoxin reductase 1 (TXNRD1; Fig. 4D). TXNRD1 is a selenoprotein that plays a central role in maintaining cellular redox homeostasis through the thioredoxin system (Nguyen et al 2006). siRNA-mediated YAP/TAZ knockdown reduced TXNRD1 protein levels (Fig. 4E). To substantiate this regulation, we conducted a JASPAR analysis, which revealed a predicted TEAD-binding motif in the TXNRD1 promoter region (Fig. 4F). CUT&RUN with a pan-TEAD antibody, followed by quantitative polymerase chain reaction, confirmed TEAD occupancy at this locus (Fig. 4G), demonstrating the transcriptional regulation of YAP/TAZ on TXNRD1. Moreover, siRNA-mediated TXNRD1 knockdown decreased the sensitivity of DOK cells to ferroptosis induction (Fig. 4H), while it elevated the intracellular redox capability as indicated by increased reduced glutathione (GSH) to oxidized glutathione (GSSG) levels (Fig. 4I). TXNRD1 expression was consistently increased in OLK tissues as compared with normal oral mucosa (Fig. 4J). We therefore concluded that TXNRD1 is a critical regulator of ferroptosis sensitivity in OLK under YAP/TAZ-TEAD control. Furthermore, since ferroptosis is driven by the peroxidation of polyunsaturated fatty acid (PUFA)–containing phospholipids and lipid metabolism dysregulation was observed in OLK, we treated DOK cells with additional PUFA docosahexaenoic acid (DHA) in combination with erastin. Remarkably, this combination produced a pronounced synergistic lethal effect (Fig. 4K).
Ferroptosis Induction Has Therapeutic Potential for OLK
Although established ferroptosis inducers such as erastin and RSL3 are highly potent, none are currently clinically available. Applying a drug-repositioning approach, we chose sulfasalazine (SAS), a clinically approved anti-inflammatory agent, as a potent inducer of ferroptosis via inhibiting the system xc− (Stockwell 2022). As expected, SAS was more effective in cells exhibiting YAP/TAZ activation or high TXNRD1 expression (Fig. 5A). When combined with DHA, SAS demonstrated an enhanced synergistic effect, with a ZIP score of 13.77 (Fig. 5B, C). Increased ferrous ion and lipid peroxidation levels confirmed that ferroptosis was triggered during the initiation phase of the combined treatment, and it remained the predominant mode of cell death over necrosis and apoptosis in the later phase (Fig. 5D, E; Appendix Fig. 5A, B).
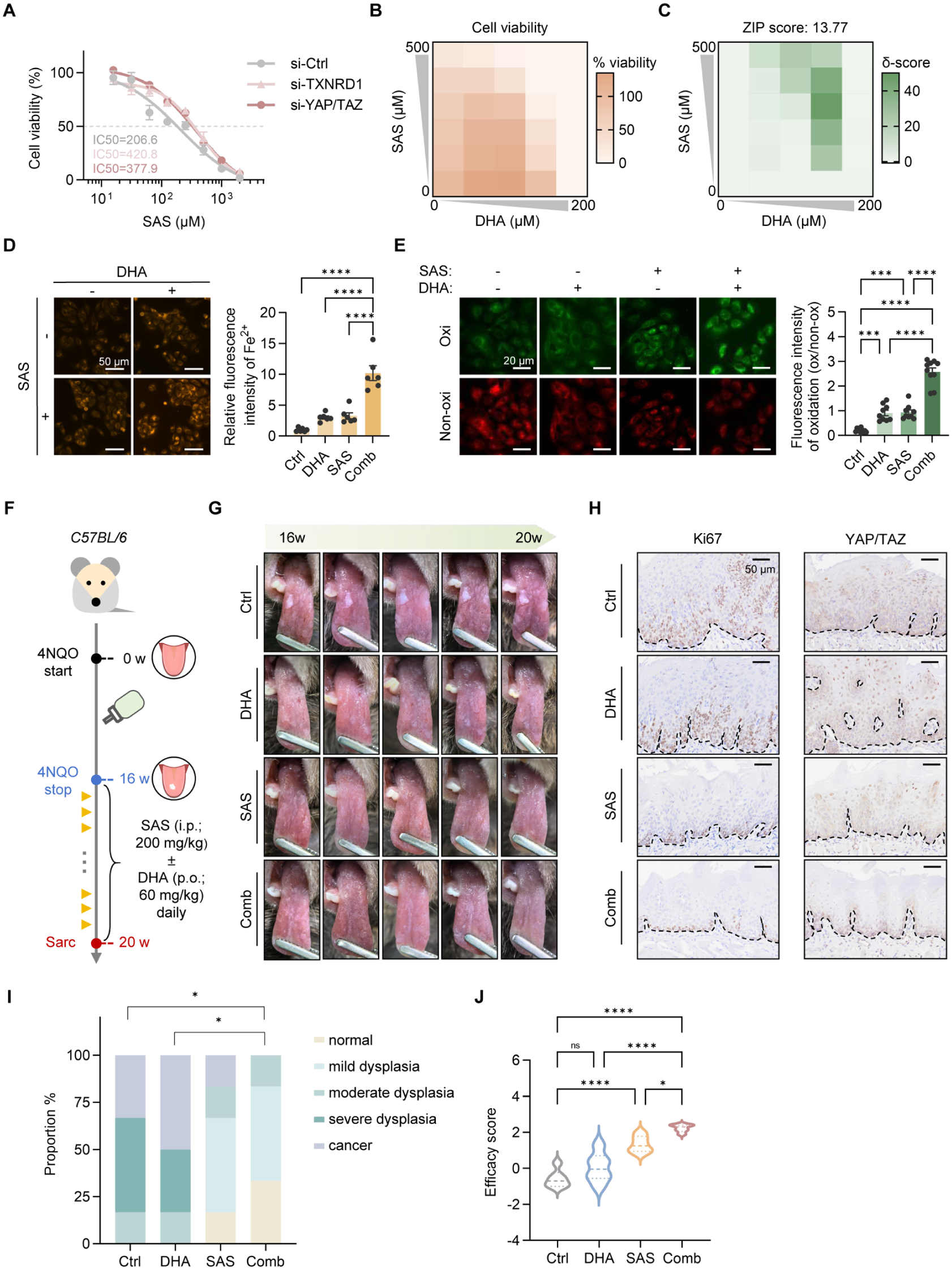
Efficacy validation of sulfasalazine (SAS) and docosahexaenoic acid (DHA) in preclinical models. (
To evaluate the efficacy of this combination strategy, an OLK mouse model was generated by 4-NQO exposure, followed by treatment with SAS, DHA, or their combination (Fig. 5F). SAS alone partially alleviated lesions, reducing epithelial dysplasia and Ki67 expression. In contrast, DHA monotherapy showed minimal benefit, with persistent dysplasia and elevated Ki67 levels. Notably, combined SAS and DHA treatment markedly improved outcomes, as evidenced by reduced lesion area, smoother mucosa, decreased dysplasia, significantly lower Ki67 expression, and more restoration of normal epithelial architecture along with a normalized YAP/TAZ expression pattern (Fig. 5G–I, Appendix Fig. 5C–E). 4HNE staining confirmed the enhanced lipid peroxidation following SAS treatment, while the addition of DHA further eliminated susceptible cells, leading to an overall reduction in 4HNE levels (Appendix Fig. 5F). No apparent toxicity was observed (Appendix Fig. 5G). Quantitative scoring based on clinical and histopathologic manifestations demonstrated the highest therapeutic efficacy in the combination group (Fig. 5J). Collectively, these findings provide the strong therapeutic potential of SAS and DHA combination therapy for the treatment of OLK.
Discussion
OLK faces notable challenges in clinical management, since current therapies provide unsatisfactory curative benefit and insufficient prevention of MT. The incomplete understanding of its etiology and molecular mechanisms complicates treatment and prognosis. Here, we identify that YAP/TAZ overactivation is strongly associated with disrupted epithelial homeostasis in OLK, contributing to expansion of basal-like cell populations. Notably, aberrant YAP/TAZ activity also confers a collateral vulnerability to ferroptosis, making ferroptosis induction a targetable and promising strategy for OLK treatment and MT prevention.
YAP/TAZ overactivation is involved throughout the progression from epithelial architectural disruption to MT. In normal stratified oral mucosa, epithelial homeostasis is maintained by basal stem-like/progenitor cells that undergo precisely regulated proliferation and differentiation, a process governed by multiple signaling pathways, with YAP/TAZ acting as the master regulators (Lian et al 2010; Zhang et al 2011; Varelas 2014). Under physiologic conditions, YAP/TAZ activation is largely confined to the basal layer through FAK-mediated mechanotransduction from integrin–basement membrane adhesion (Peng et al 2025). However, persistent carcinogenic stimuli can lead to YAP/TAZ hyperactivation beyond the basal layer, ultimately driving tumor initiation (Zanconato et al 2016; Maglic et al 2018; Omori et al 2020). Therefore, aberrant YAP/TAZ activity in OLKs puts them at high risk of MT. Our integrated bioinformatics and functional analyses identify this escape from basal confinement as a key molecular event in OLK, linking YAP/TAZ activation to excessive proliferation and malignant phenotypes.
Although genetic inhibition of YAP/TAZ attenuated OLK progression in our transgenic mice models, direct pharmacologic targeting remains clinically impractical. The YAP transcriptional activation domain lacks well-defined druggable structural pockets, and most current approaches aim to disrupt YAP-TEAD interactions instead (Liu-Chittenden et al 2012; Yuan et al 2025) but with limited clinical applicability. Moreover, as a central integrator of diverse cellular signals, YAP/TAZ regulate broad transcriptional programs governing proliferation, differentiation, and survival. This pleiotropy presents a challenge for therapeutic intervention, as global inhibition of YAP/TAZ may elicit unwanted systemic homeostasis disruption and severe off-target effect toxicity (Driskill and Pan 2023); it may even paradoxically activate prosurvival pathways such as the SOX4/PI3K/AKT axis (Sun et al 2022). Importantly, YAP/TAZ hyperactivation is double-edged: while promoting cell survival, it also creates intrinsic vulnerabilities (Yang et al 2018; Wu et al 2019). Consistent with this concept, we observed heightened ferroptosis sensitivity in OLK, indicating that YAP/TAZ-driven lesions harbor an exploitable vulnerability that may offer an alternative approach to address otherwise undruggable YAP/TAZ signaling.
Mechanistically, ferroptosis regulation is highly context dependent. In epithelium, YAP activation has been reported to upregulate ferroptosis-related genes such as acyl-CoA synthetase long chain family membner 4 (ACSL4) and transferrin receptor (Wu et al 2019). Here, we identify TXNRD1 as a novel YAP/TAZ-controlled modulator of ferroptosis in OLK. TXNRD1 is a key antioxidant enzyme within the thioredoxin system, whereas glutathione peroxidase (GPX4) serves as the central lipid peroxide detoxifier. Prior studies have shown that knocking down TXNRD1 in cancer cells can increase GPX4 protein levels (Cai et al 2020). We consistently found that TXNRD1 depletion enhanced GSH-mediated redox capacity in epithelial cells. This observation suggests a functional trade-off between TXNRD1 and GPX4, potentially due to their shared dependence on selenium and NADPH. Both enzymes contain selenocysteine residue at their redox-active sites, implying that reduced TXNRD1 expression may increase selenium availability for GPX4 synthesis. In addition, NADPH donates electrons to TXNRD1 to maintain thioredoxin in a reduced state, a process essential for peroxide detoxification, and to GSH reductase to regenerate GSH from oxidized GSSG. Consequently, TXNRD1 and GPX4 may compete for limited NADPH to maintain redox balance under oxidative stress, resulting in an altered GSH/GSSG ratio. Collectively, our study demonstrates that YAP/TAZ are involved in regulating the redox system within the ferroptosis-associated pathway in epithelium. With previous reports finding that YAP/TAZ participate in ferroptosis regulation via lipid and iron metabolism genes (Wu et al 2019), these findings underscore the critical role of YAP/TAZ in controlling epithelial ferroptosis.
Ferroptosis has emerged as a promising therapeutic target for diverse diseases, including cancer and neurodegenerative disorders (Stockwell 2022). A common strategy is disruption of the solute carrier family 7 member 11 (SLC7A11)-GSH-GPX4 axis (Badgley et al 2020). Although small-molecule inducers (e.g., erastin, RSL3, FIN56) show strong preclinical efficacy, challenges in solubility, bioavailability, and toxicity limit their clinical application. Rather than developing novel compounds, we explored a drug-repurposing strategy using SAS, an approved anti-inflammatory agent recently shown to target system xc− in cancer (Chen et al 2021), and demonstrated its therapeutic efficacy in OLK models. Malignant cells frequently adapt lipid metabolism to survive under metabolic stress, which may alter their susceptibility to ferroptosis due to heightened metabolic activity and elevated reactive oxygen species burden. Notably, we observed heightened lipid metabolic activities and increased lipid peroxidation in OLK, indicating its inherent susceptibility to ferroptosis. Given that PUFA-containing phospholipids are essential for ferroptotic membrane damage (Yang et al 2016; Pope and Dixon 2023), we leveraged DHA, a highly oxidation-prone ω-3 PUFA with 6 double bonds in its structure, which is widely recommended as a dietary supplement. In our findings, DHA alone showed limited therapeutic efficacy; however, by creating a more oxidation-prone environment, it sensitized cells to redox imbalance, thereby amplifying the therapeutic effect when combined with SAS. These findings establish ferroptosis induction, coupled with nutritional intervention, as a feasible therapeutic strategy for OLK with great potential for accelerating clinical translation.
In conclusion, this study provides advanced insights into OLK pathogenesis and highlights the dual role of YAP/TAZ in OLK. On the one hand, aberrant YAP/TAZ activity is involved in disrupted epithelial homeostasis, represented by basal-like population expansion, and it promotes OLK progression. On the other, this hyperactivation simultaneously creates a ferroptosis vulnerability via TEAD-TXNRD1 in these cells. By exploiting this intrinsic liability, we identify ferroptosis induction with clinically approved SAS and DHA as a potentially actionable and translational strategy for improving OLK management.
Author Contributions
D. Yang, contributed to conception and design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; L. Zhang, contributed to acquisition, analysis, and interpretation, drafted and critically revised the manuscript; T. Zhou, contributed to conception, data acquisition, critically revised the manuscript; Q. Yuan, J. Zhang, contributed to data acquisition, critically revised the manuscript; X. Xia, contributed to design, critically revised the manuscript; H. Dan, contributed to conception, critically revised the manuscript; X. Feng, contributed to conception and design, critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261451778 – Supplemental material for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability
Supplemental material, sj-docx-1-jdr-10.1177_00220345261451778 for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability by D. Yang, L. Zhang, T. Zhou, Q. Yuan, J. Zhang, X. Xia, H. Dan and X. Feng in Journal of Dental Research
Supplemental Material
sj-xlsx-2-jdr-10.1177_00220345261451778 – Supplemental material for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability
Supplemental material, sj-xlsx-2-jdr-10.1177_00220345261451778 for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability by D. Yang, L. Zhang, T. Zhou, Q. Yuan, J. Zhang, X. Xia, H. Dan and X. Feng in Journal of Dental Research
Supplemental Material
sj-xlsx-3-jdr-10.1177_00220345261451778 – Supplemental material for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability
Supplemental material, sj-xlsx-3-jdr-10.1177_00220345261451778 for YAP/TAZ Drive Oral Leukoplakia Progression and Confer Ferroptosis Vulnerability by D. Yang, L. Zhang, T. Zhou, Q. Yuan, J. Zhang, X. Xia, H. Dan and X. Feng in Journal of Dental Research
Footnotes
Acknowledgements
We thank Professor Faxing Yu for kindly providing SuperHippo transgenic mice and appreciate the contributions of all clinical participants. We also thank Ning Ji (State Key Laboratory of Oral Diseases, Sichuan University) for excellent assistance with microscopic imaging.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundations of China (grants 82170971, 82373187, 82571090, 82370965), Sichuan Science and Technology Program (2025NSFJQ0062), and Fund of State Key Laboratory of Oral Diseases (SKLOD-2025KP009).
Data Availability Statement
The data generated in this study are available within the article and its Appendix. Bioinformatics datasets were obtained from the GEO database (scRNA-seq: GSE181919; bulk RNA-seq: GSE227919 and GSE136876), and the code used for analyses is available on GitHub (![]() ). Appendix Tables 1 to 4 contain bioinformatics analysis results. Appendix data files provide the research data for all reported assays (immunohistochemical quantification, cell viability assays, quantitative polymerase chain reaction assays, detection assays, and animal studies) corresponding to the figures cited in the article and Appendix.
). Appendix Tables 1 to 4 contain bioinformatics analysis results. Appendix data files provide the research data for all reported assays (immunohistochemical quantification, cell viability assays, quantitative polymerase chain reaction assays, detection assays, and animal studies) corresponding to the figures cited in the article and Appendix.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.