
Abstract
Incomplete regeneration of the junctional epithelium (JE) following periodontal therapy is associated with persistent alveolar bone resorption and disease recurrence. While pathogen clearance is essential, the host-intrinsic mechanisms dictating epithelial recovery remain obscure. Here, we reveal that Porphyromonas gingivalis challenge induces an aberrant jamming-like phase transition in the epithelium during the early recovery window, mechanically restricting the epithelial remodeling required for proper JE reconstruction. Through a genome-wide CRISPR-Cas9 screen, we identified Dynein Light Chain LC8-Type 1 (DYNLL1) as a central mediator of this pathological transition. Mechanistically, DYNLL1 regulates the abundance and membrane localization of the junction-associated protein JUP, whose overaccumulation strengthens intercellular adhesion and restricts epithelial motility. Partial DYNLL1 deficiency not only reduces junctional rigidity but also enhances Wnt/β-catenin signaling, thereby preserving a more dynamically responsive epithelial state conducive to repair. In a murine periodontitis model, Dynll1 haploinsufficiency facilitated the restoration of JE morphology, improved barrier function, and mitigated bone loss. These findings suggest that modulating epithelial biophysical properties may complement antimicrobial therapies to enhance periodontal healing. Overall, this work provides novel insights into host-intrinsic mechanisms that contribute to impaired periodontal repair and identifies potential therapeutic targets for early-stage intervention.
Introduction
Periodontitis is characterized by the progressive loss of periodontal attachment and supporting tissues. Durable periodontal repair requires reestablishment of a functional junctional epithelial (JE) seal on the tooth surface (Li et al 2020). The JE is a unique structure with wide intercellular spaces, sparse junctional complexes, and rapid turnover (Kitsuki et al 2020; Yuan et al 2021). Compared to the oral gingival epithelium, the JE constitutively expresses higher levels of proliferation and migration drivers such as epidermal growth factor and lower levels of intercellular adhesion proteins, including E-cadherin (Lee et al 2016; Fischer and Aparicio 2022). This dynamic and relatively weakly adhesive architecture may be important for maintaining the functional cell–tooth attachment within the complex gingival crevice microenvironment.
However, this homeostasis is disrupted in the periodontitis microenvironment. Porphyromonas gingivalis (P. gingivalis), a keystone pathogen in periodontitis, exploits host nutrients to establish intracellular persistence (Andrian et al 2006; Chopra et al 2020). While the epithelium remains viable and chronic inflammation persists, a functional epithelial seal fails to reestablish. Given that epithelial migration at chronic wound edges is often severely limited (Pastar et al 2014; Theocharidis et al 2022), we hypothesized that P. gingivalis impairs epithelial migratory plasticity. Our data show that during early recovery, epithelial populations challenged by P. gingivalis exhibit proliferative but confined, overcrowded structures reminiscent of epithelial jamming—a phase transition from a fluid-like to a solid-like state that restricts motility (Atia et al 2018). These findings suggest that P. gingivalis may induce a jamming-like state during early periodontal inflammation or posttreatment recovery, mechanically hindering JE remodeling and functional reconstruction.
Susceptibility to periodontitis and repair efficacy vary significantly among individuals, partly attributable to host genetics (Nibali et al 2017; Öztürk and Ada 2022), suggesting that intrinsic programs may modulate epithelial responses to bacterial stress. Hypothesizing that specific regulators promote this epithelial jamming-like phase transition, we performed a genome-wide CRISPR-Cas9 screen (Wang et al 2021; Chen et al 2025) to identify host genes whose depletion confers a fitness advantage against bacterial pressure.
Here, we identified Dynein Light Chain LC8-Type 1 (DYNLL1, also known as LC8) as a critical modulator of the P. gingivalis–induced epithelial jamming-like phase transition. Mechanistically, DYNLL1 regulates epithelial biophysical properties by controlling the abundance and subcellular localization of JUP (also known as γ-catenin). P. gingivalis challenge disrupts DYNLL1–JUP interaction, triggering a JUP-dependent excessive accumulation of adherens junctions that mechanically locks the tissue in a solid-like state. Conversely, partial DYNLL1 depletion reduces junction-associated JUP and promotes Wnt signaling, shifting the epithelium toward a more dynamically responsive state that may favor collective migration and lessen susceptibility to the jamming-like phase transition. In a murine periodontitis recovery model, Dynll1 haploinsufficiency facilitates the reconstruction of JE morphology and function and alleviates alveolar bone loss.
Our study identifies the DYNLL1/JUP complex as a central molecular switch governing the epithelial jamming-like phase transition and suggests that modulating the biophysical properties of the epithelium could represent a promising strategy to enhance periodontal repair in periodontitis.
Materials and Methods
Key experimental approaches included the establishment of stable cell lines, P. gingivalis challenge, epithelial cell jamming-like phase transition assay, time-lapse high-content imaging, genome-wide CRISPR-Cas9 knockout screening, bulk RNA sequencing, immunoprecipitation–mass spectrometry (IP-MS), co-immunoprecipitation, proximity ligation assay, mouse model of periodontitis, and immunofluorescence staining. Detailed protocols are provided in the Appendix. All animal studies followed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) 2.0 guidelines.
Results
P. gingivalis Challenge Induces a Jamming-Like State during the Recovery Phase That Arrests Epithelial Collective Dynamics and Impedes Repair
To investigate whether transient P. gingivalis challenge induces an epithelial jamming-like phase transition, we established a compartmentalized challenge–recovery model spatially separating P. gingivalis–challenged cells (Pg+ region) from unchallenged bystander cells (Pg− region) (Fig. 1A). Human immortalized oral epithelial cells (HIOECs) in the Pg+ region initially retracted and subsequently formed densely packed clusters, accompanied by sustained proliferation that seemingly bypassed contact inhibition signals (Fig. 1B, Appendix Fig. 1A). This population acted as a physical barrier. Consequently, bystander cells in the Pg− region failed to migrate past this boundary, accumulating against the static challenged front and leading to localized overcrowding. Both regions eventually progressed to a multilayered state, while a clear boundary remained between them.
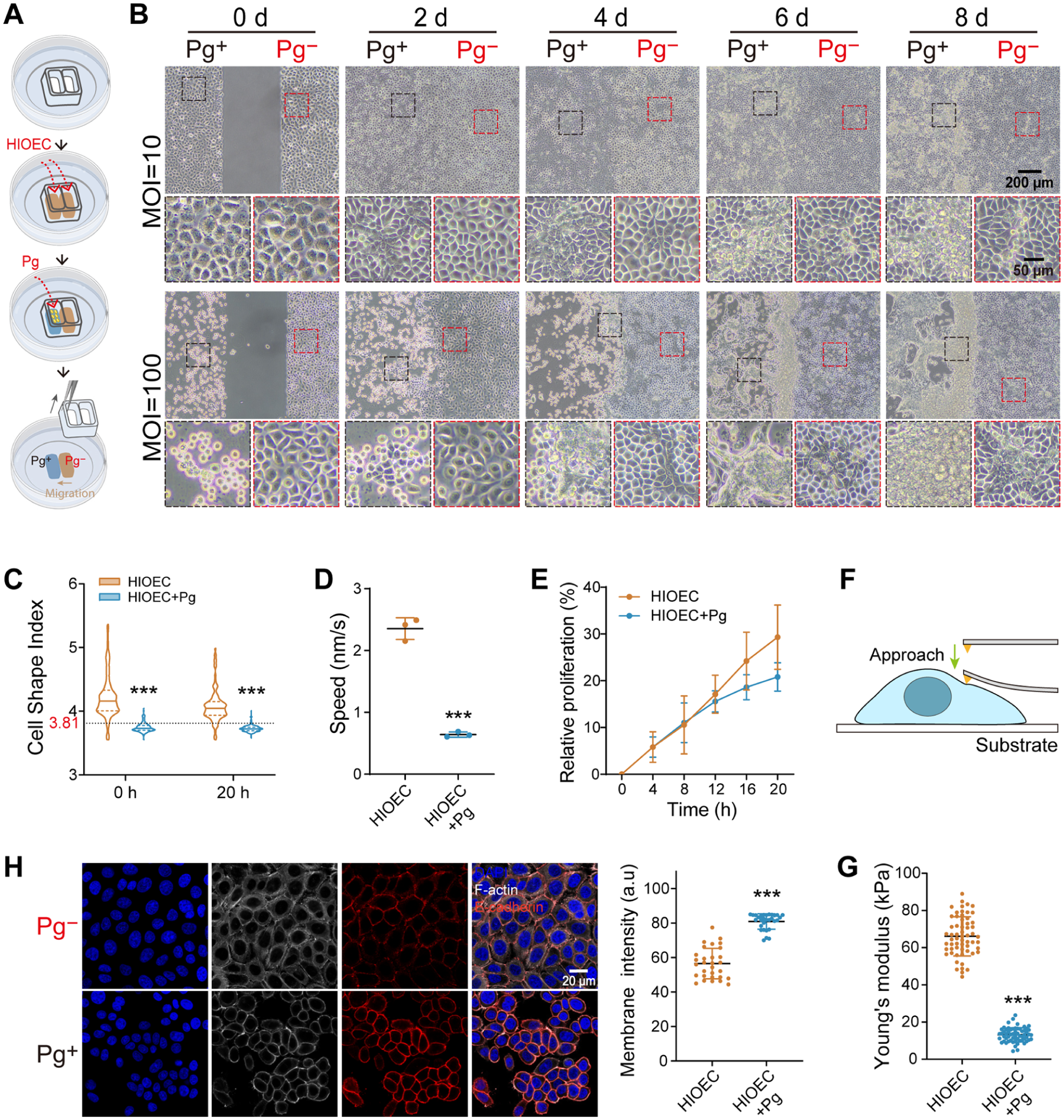
Transient Porphyromonas gingivalis (P. gingivalis) challenge induces an epithelial jamming-like state during recovery. (
Live-cell imaging (Appendix Videos 1–2) confirmed that during the postchallenge recovery phase, the cell shape index (
To investigate the biomechanical properties underlying this arrest, we measured cell stiffness using atomic force microscopy (AFM) (Fig. 1F). Accompanying this jamming-like state, challenged cells exhibited a significant decrease in Young’s modulus, indicating a softening of the cell body (Fig. 1G) (Wagh 2008; Pajic-Lijakovic and Milivojevic 2022). Immunofluorescence staining revealed that E-cadherin accumulated excessively at intercellular junctions (Fig. 1H), which was corroborated by Western blot analysis showing increased levels of both total and membrane-associated E-cadherin protein during the recovery phase (Appendix Fig. 2). Phosphorylated myosin light chain (pMLC) redistributed from polarized stress fibers to the entire cell cortex (Appendix Fig. 1B). These results indicate a loss of the polarized contractility required for active migration and support the transition toward a jamming-like state.
These data demonstrate that transient P. gingivalis challenge alters epithelial cell biophysical properties during postchallenge recovery, driving the tissue into a jamming-like state and mechanically impeding repair by adjacent unchallenged cells.
Genome-wide CRISPR Screening Identifies DYNLL1 as a Key Mediator of the P. gingivalis–Induced Epithelial Jamming-Like Phase Transition during the Recovery Phase
To identify host factors governing the epithelial jamming-like phase transition following P. gingivalis challenge, we performed a genome-wide CRISPR-Cas9 knockout screen (Fig. 2A). To establish a robust selection pressure (Appendix Fig. 3A–C), the mutant library was subjected to a P. gingivalis challenge (multiplicity of infection = 60), followed by a recovery phase. This dual-pressure strategy specifically enriched for survivors—cells that not only resisted challenge-induced detachment (Appendix Fig. 3D) but also resumed postchallenge outgrowth. Sequencing of this surviving population and subsequent MAGeCK analysis identified a set of genes with RRA P < 0.05 (Appendix Fig. 4A).
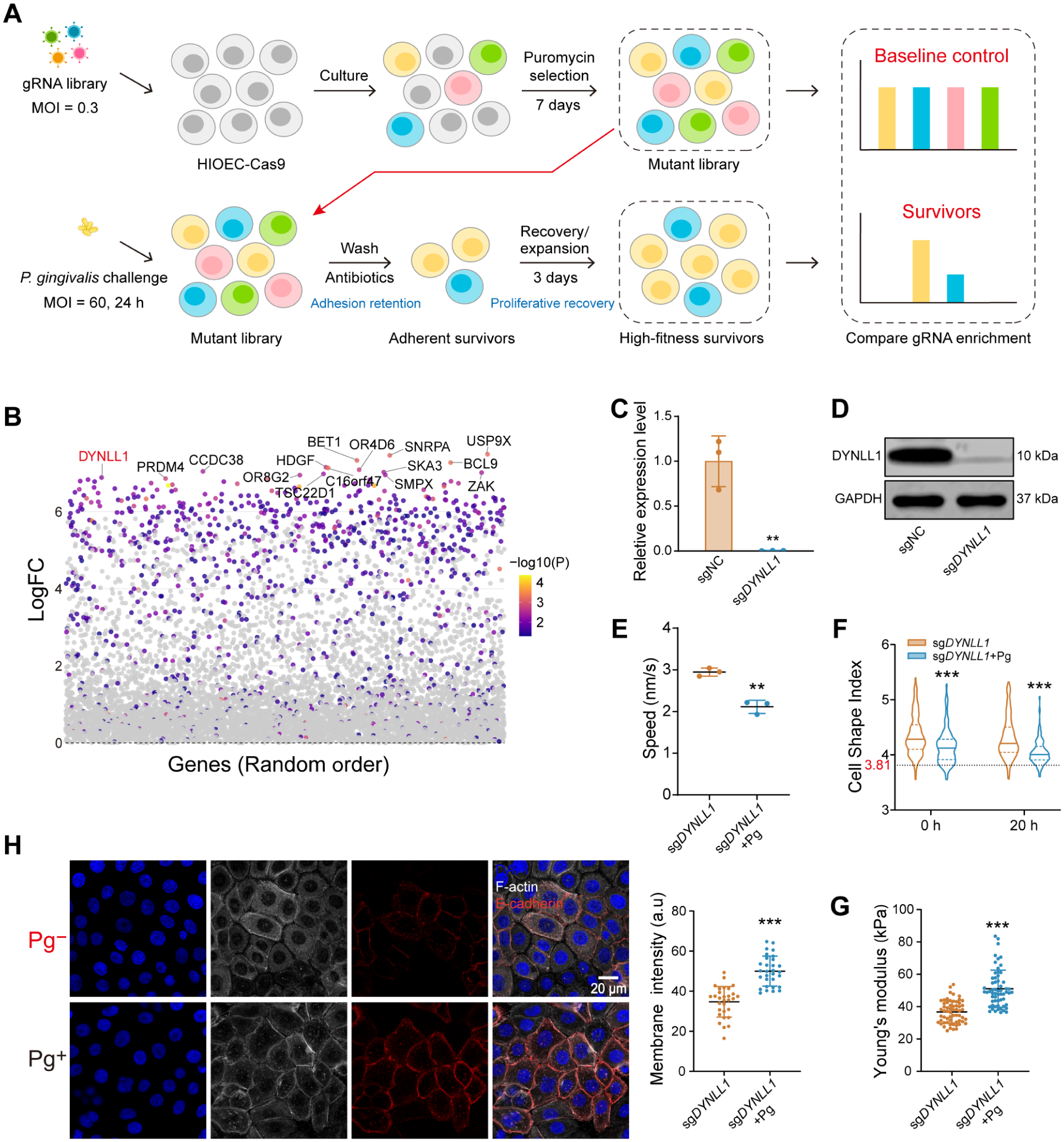
Genome-wide CRISPR screening and functional validation identify DYNLL1 as a regulator of postchallenge epithelial jamming-like dynamics. (
By cross-referencing the top 15 LFC-ranked candidates (Fig. 2B) with single-cell RNA sequencing (scRNA-seq) datasets from human gingival tissue in healthy and periodontitis states (Zhang et al 2026), we found that DYNLL1 expression was notably upregulated in diseased epithelial clusters (Appendix Fig. 4B), suggesting disease-associated upregulation of DYNLL1 in periodontal epithelium. Due to the lethality of a complete knockout, we generated a stable heterozygous knockout cell line (sgDYNLL1) for subsequent functional validation (Fig. 2C, D, Appendix Fig. 5). Nontargeting cells (sgNC, equivalent to the HIOECs characterized in Fig. 1C–H) served as controls.
Functionally, DYNLL1 depletion intrinsically enhanced cell proliferation (Appendix Fig. 6A) and migration, as evidenced by accelerated wound healing and a higher lamellipodia index (Appendix Fig. 6B–E). While P. gingivalis challenge significantly arrested sgNC during the recovery phase, sgDYNLL1 maintained a migration velocity comparable to unchallenged sgNC (~2 nm/s) and exhibited a shape index consistently above the critical jamming threshold of 3.81 (Fig. 2E, F, Appendix Fig. 6F, Appendix Videos 3–4). Biomechanically, challenged sgDYNLL1 resisted challenge-induced cellular softening, maintaining stiffness levels resembling those of healthy sgNC (Figs. 1G, 2G). While challenge triggered E-cadherin upregulation during the recovery phase, junctional E-cadherin accumulation in sgDYNLL1 cells approximated that of healthy sgNC rather than the excessive accumulation seen in challenged sgNC cells (Fig. 2H). The distribution of pMLC in challenged sgDYNLL1 retained polarity and preserved the contractile machinery essential for active migration (Appendix Fig. 1C).
Our findings identify DYNLL1 as a key mediator and demonstrate that its partial depletion confers resistance to the P. gingivalis–induced epithelial jamming-like phase transition during the postchallenge recovery phase.
DYNLL1 Regulates Epithelial Dynamics by Controlling the Abundance and Nuclear Localization of JUP
To clarify the molecular mechanisms by which DYNLL1 regulates epithelial dynamics, we identified the differentially expressed genes (DEGs) between sgNC and sgDYNLL1 cells alongside DYNLL1-binding proteins (the interactome) using transcriptomic and IP-MS analyses, respectively (Fig. 3A, B). Gene Ontology (GO) analysis revealed enrichment in terms associated with protein binding, vesicle transport, and cell cycle regulation (Appendix Fig. 7A–C). Additionally, a subset of candidates was significantly enriched in GO terms related to cell migration, intercellular junctions, and defense responses.
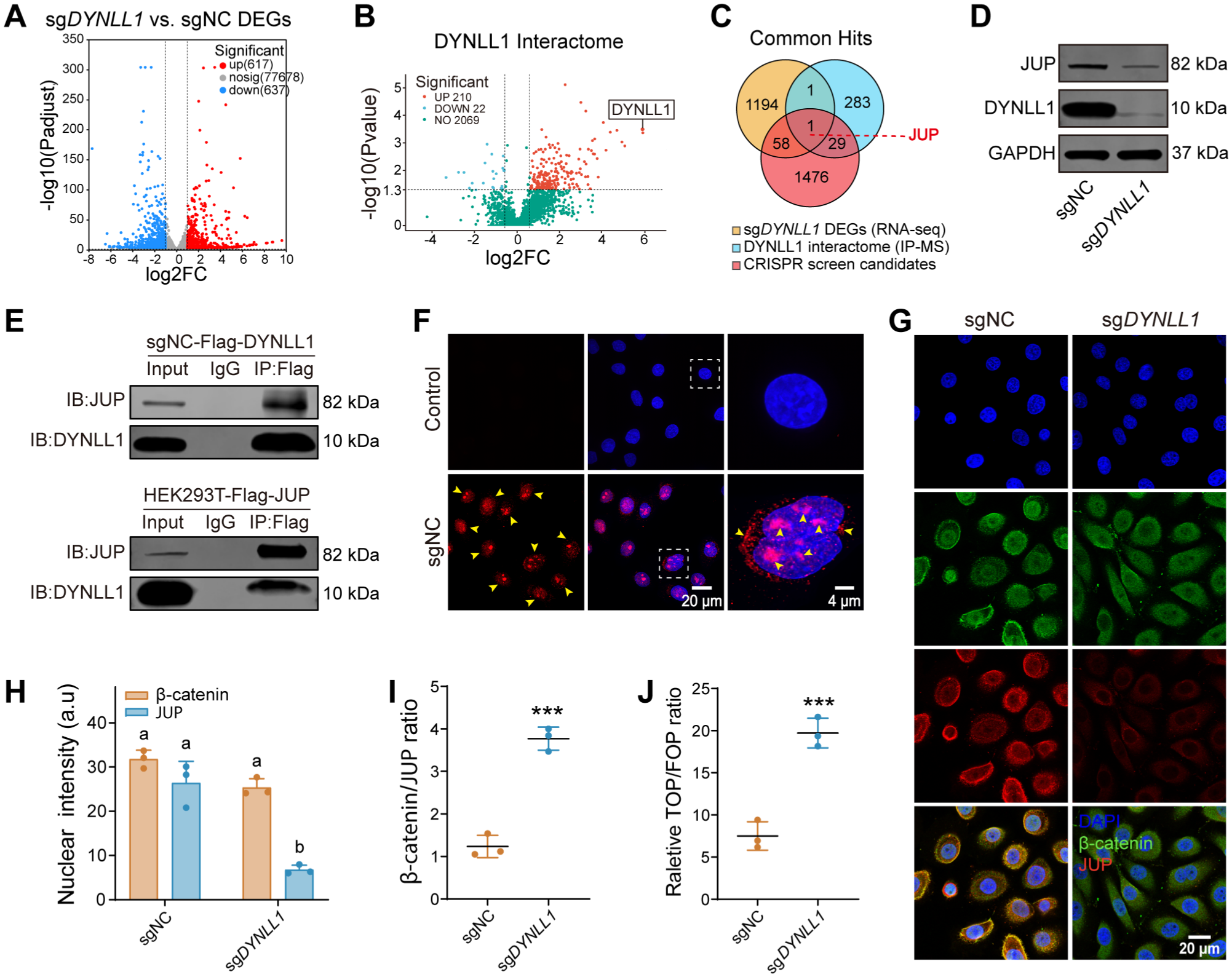
Multiomics integration reveals that DYNLL1 modulates Wnt signaling via the regulation of JUP abundance and nuclear localization. (
We identified JUP as an important regulatory node by intersecting the transcriptomic DEGs, the DYNLL1 interactome, and CRISPR positive-selection candidates (Fig. 3C), suggesting a functional link between DYNLL1 and JUP following P. gingivalis challenge. We confirmed that DYNLL1 depletion significantly reduced JUP protein levels (Fig. 3D). Reciprocal co-immunoprecipitation (co-IP) and proximity ligation assays (PLAs) verified a physical interaction between DYNLL1 and JUP (Fig. 3E, F, Appendix Fig. 8A–C). Signals from the PLAs were predominantly localized to the perinuclear and nuclear regions, suggesting that DYNLL1 is associated with the nuclear localization of JUP.
Since JUP is known to compete with β-catenin for TCF binding to drive distinct downstream effects (Charpentier et al 2000; Miravet et al 2002), we examined their subcellular distribution. In sgNC cells, JUP exhibited clear nuclear localization. In contrast, DYNLL1 depletion not only downregulated overall JUP abundance but also caused its retention in the cytoplasm. Nuclear β-catenin levels remained stable between the two groups (Fig. 3G, H). This significantly increased the ratio of nuclear β-catenin to JUP in sgDYNLL1 cells, suggesting relief of JUP-mediated inhibition of Wnt signaling (Fig. 3I). TOPFlash dual-luciferase assays further demonstrated stronger activation of the Wnt/β-catenin pathway in sgDYNLL1 cells (Fig. 3J). These results indicate that DYNLL1 is essential for maintaining JUP abundance and nuclear localization. Its depletion triggers JUP downregulation and cytoplasmic retention, thereby relieving JUP-mediated restraint on β-catenin–driven transcription and shifting epithelial cells toward a more dynamically responsive state.
P. gingivalis Challenge Disrupts the DYNLL1/JUP Complex and Promotes Junctional JUP Accumulation
We found that the regulatory interaction between DYNLL1 and JUP is sensitive to P. gingivalis challenge. The challenge significantly disrupted the DYNLL1/JUP complex, as evidenced by the loss of their physical association in reciprocal co-IP assays (Fig. 4A). Under homeostatic conditions, DYNLL1 and JUP exhibit a dual distribution in the nucleus and cytoplasm. However, following P. gingivalis challenge, a marked redistribution of JUP was observed during the recovery phase, characterized by pronounced nuclear depletion and substantial accumulation in the cytoplasm and at the cell membrane (Fig. 4B–F). This significant membrane enrichment of JUP is consistent with the excessive accumulation of E-cadherin observed during the postchallenge recovery phase (Fig. 1H), given that JUP contributes to the stability of E-cadherin (Ding et al 2025).
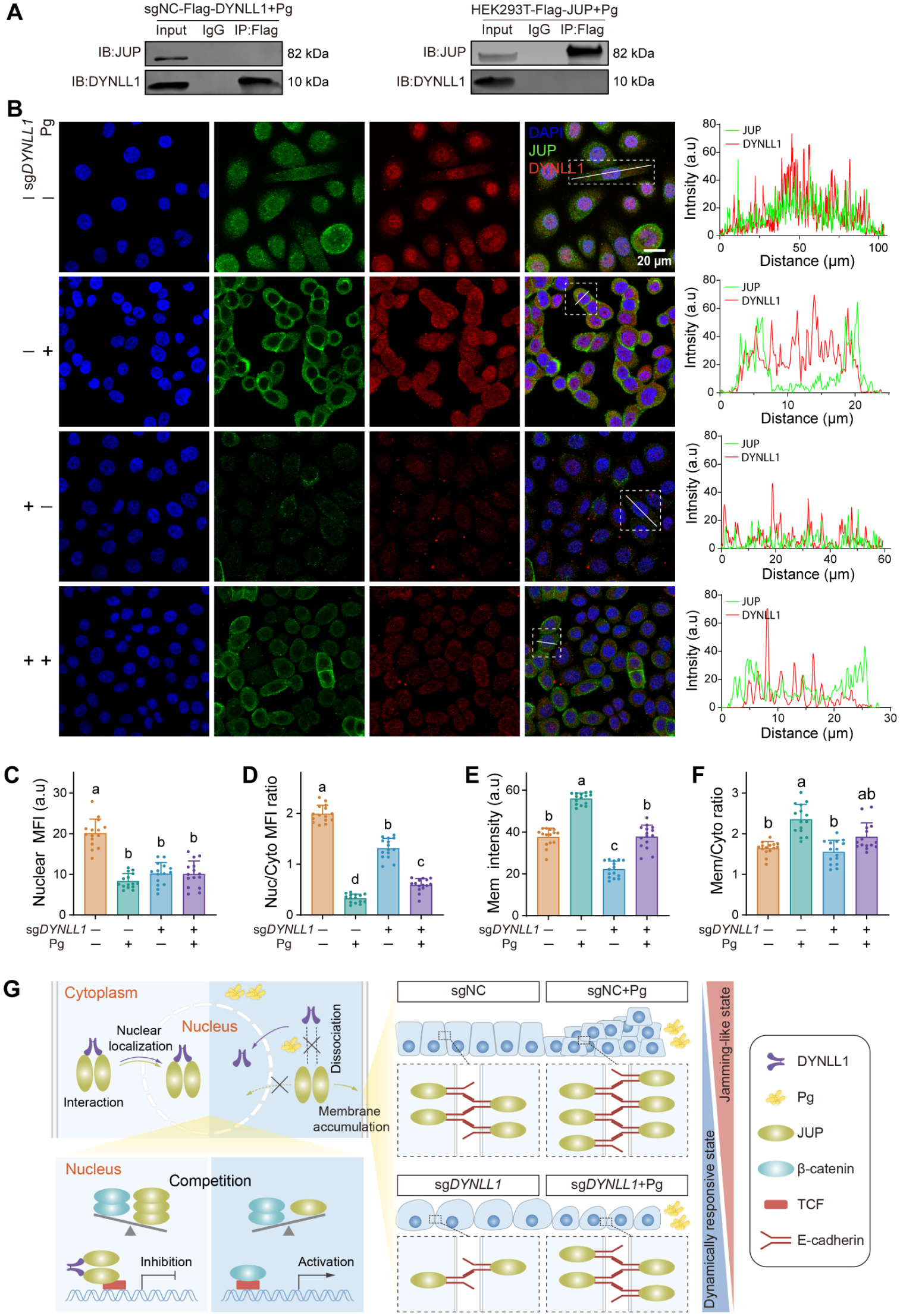
Porphyromonas gingivalis challenge disrupts the DYNLL1/JUP complex and promotes junctional JUP accumulation during recovery. (
Collectively, these results indicate that P. gingivalis challenge perturbs DYNLL1-dependent regulation of JUP, promoting JUP redistribution toward the cell membrane and excessive accumulation of adherens junction components, thereby favoring the pathological phase transition (Fig. 4G).
DYNLL1 Insufficiency Resists Jamming-Like Epithelial Remodeling and Promotes Periodontal Repair In Vivo
To evaluate the physiological relevance of our findings in vivo, we generated Dynll1 heterozygous knockout mice (Dynll1+/−), as homozygous deletion is lethal (Appendix Fig. 9A, B). Dynll1+/− mice exhibited reduced JUP protein levels without altering the expression patterns of the JE marker ODAM (Liang et al 2026) and the hemidesmosome components BP180 and Laminin5/Laminin332, indicating that moderate DYNLL1 depletion does not compromise periodontal integrity (Appendix Fig. 9C).
Following the combined ligature + P. gingivalis periodontitis/recovery protocol (Fig. 5A), the epithelium of periodontitis-induced wild-type mice (WT_P) exhibited excessive JUP and E-cadherin accumulation at intercellular junctions alongside significantly increased cell density (Fig. 5B–G). Furthermore, the long axis of these cells was preferentially aligned parallel to the basement membrane, indicating disrupted apicocoronal orientation associated with JE remodeling (Fig. 5H). Despite the high cell density, the proportion of Ki67+ proliferative cells in the WT_P basal layer did not decrease (Appendix Fig. 10), indicating local accumulation without effective migration. In contrast, the periodontitis-induced Dynll1+/− (Dynll1+/−_P) epithelium resisted excessive junctional JUP/E-cadherin accumulation and maintained a relatively normal physiological state.
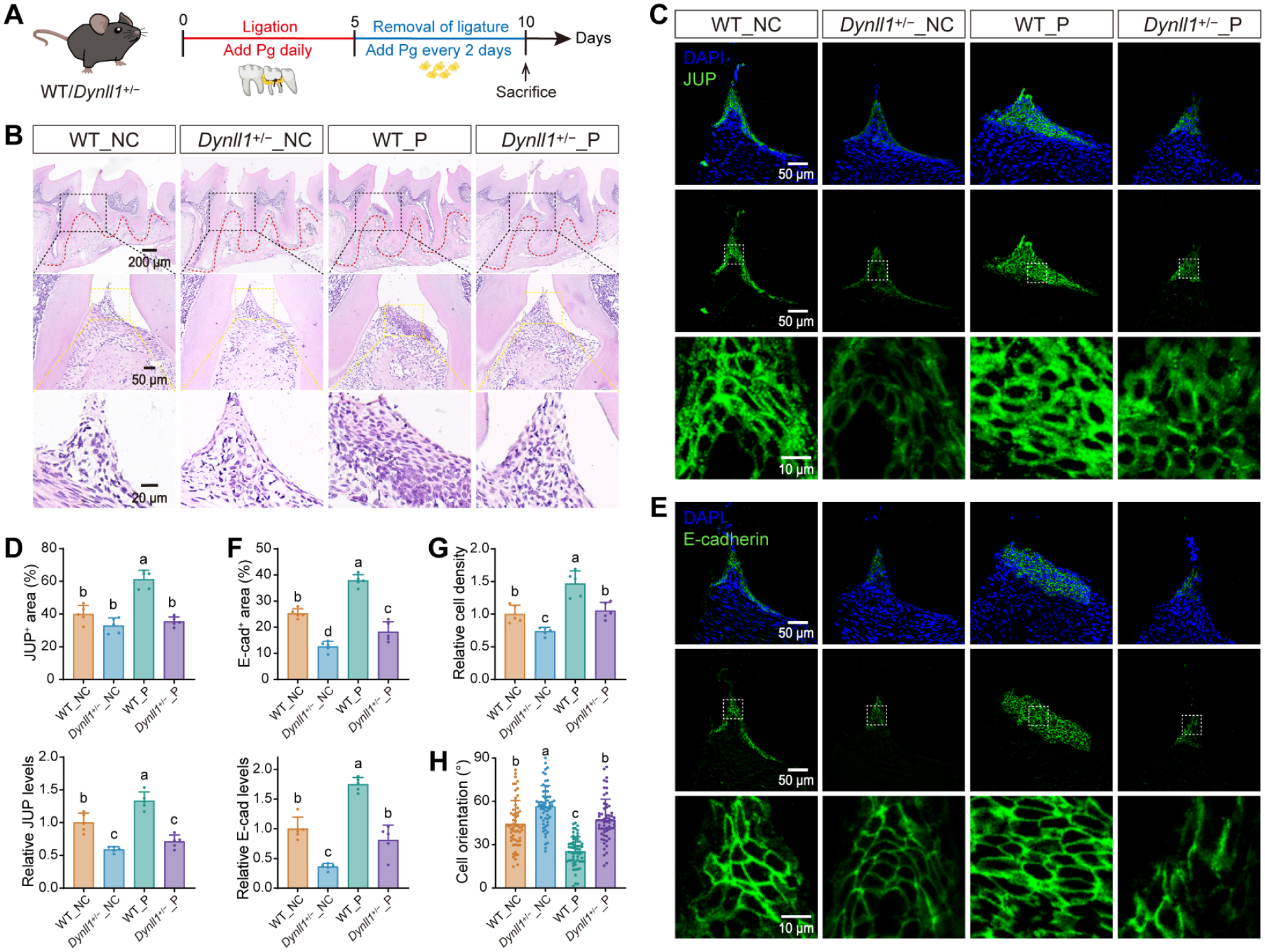
DYNLL1 insufficiency resists jamming-like epithelial remodeling in vivo. (
This aberrant jamming-like epithelial remodeling in WT_P mice was associated with impaired morphological and functional reconstruction of the JE and increased alveolar bone loss. WT_P mice exhibited a disordered distribution of ODAM, BP180, and Laminin5, reflecting a severe defect in establishing a functional cell–tooth attachment. Conversely, in the Dynll1+/−_P group, both JE morphology and function were reestablished in a more organized manner (Appendix Fig. 11). Consistent with the compromised epithelial barrier, micro–computed tomography (micro-CT) analysis revealed more severe alveolar bone loss with visible buccal fenestration-like defects in WT_P mice compared to Dynll1+/−_P mice (Appendix Fig. 12A, B). Prominent RANKL-positive signals on the bone surface confirmed heightened osteoclastic activity in the WT_P group (Appendix Fig. 12C).
To clarify the functional link between the jamming-like epithelial remodeling and bone resorption, we examined the localization of key cytokines (IL-1β and IL-6) and infiltrating immune cells (CD68 and Ly6G) (Appendix Fig. 13). In WT_P mice, these inflammatory signals were predominantly concentrated in the underlying connective tissue and near the alveolar bone. In contrast, in Dynll1+/−_P mice, they were largely confined to the epithelial compartment and were less evident near the alveolar bone.
These in vivo data indicate that, within the combined ligature + P. gingivalis periodontitis/recovery model, Dynll1 haploinsufficiency is associated with resistance to jamming-like epithelial remodeling, improved JE repair, and reduced alveolar bone loss.
Discussion
Early intervention during the initial stages of periodontitis, as well as the early healing phase following periodontal therapy, is critical for limiting tissue damage and promoting regeneration (Kinane et al 2017). However, even after effective microbial control, restoration of periodontal structures often remains incomplete, contributing to delayed healing and disease recurrence (Hajishengallis 2014). As the primary epithelial attachment directly interfacing with the tooth surface, the JE serves as the frontline barrier against subgingival bacterial invasion and is therefore central to periodontal homeostasis and posttreatment recovery. Our study focuses on this crucial early period and reveals that the epithelium undergoes an aberrant jamming-like phase transition during the recovery phase after P. gingivalis challenge. This state shares key features with classical biophysical jamming—including increased cell density, reduced cell shape index, restricted individual motility, and strengthened intercellular adhesion (Palamidessi et al 2019; Lenne and Trivedi 2022)—but arises as a challenge-induced host response rather than from physiological spatial constraints such as developmental confinement or contact inhibition. We interpret this jamming-like phase transition as an active contributing factor that mechanically restricts epithelial dynamic remodeling during the early repair phase, offering a plausible biophysical explanation for incomplete periodontal healing after bacterial challenge.
Mechanistically, we identified the DYNLL1/JUP complex as a key regulator of this pathological transition. While DYNLL1 is traditionally characterized as a light chain of the dynein motor complex essential for cytoskeletal organization and intracellular cargo transport (Williams et al 2007; He et al 2018), our data reveal a previously unrecognized role in regulating the abundance and localization of JUP. Analysis of scRNA-seq datasets from human periodontitis tissues revealed that DYNLL1 is upregulated in diseased epithelial clusters, supporting the clinical relevance of the DYNLL1/JUP complex. Upon P. gingivalis challenge, dysregulated JUP localization then promotes E-cadherin overaccumulation at adherens junctions, imposing mechanical constraints on the cells. These constraints inhibit cell polarization, cytoskeletal remodeling, and the neighbor-exchange events required for collective migration (Friedl and Gilmour 2009; Ilina et al 2020; Suh et al 2024). Concurrently, sustained proliferation within this spatially restricted environment drives local crowding, ultimately pushing the cell population beyond its critical jamming threshold.
DYNLL1 insufficiency effectively disrupts this pathological trajectory. Functionally, DYNLL1 depletion enhances cell migration capacity, indicating that sgDYNLL1 cells possess an intrinsic capacity to maintain the motility required for tissue remodeling. At the molecular level, we propose that DYNLL1 insufficiency exerts two coordinated protective effects. First, it reduces JUP availability, thereby preventing excessively dense intercellular junctions and restoring the dynamic adhesion turnover necessary for cell rearrangement. Second, it likely relieves the JUP-mediated competitive inhibition of β-catenin. Given that JUP competes with β-catenin for TCF binding, driving distinct downstream transcriptional programs, its downregulation may enhance canonical Wnt/β-catenin signaling (Kolligs et al 2000; Maeda et al 2004). While canonical Wnt signaling is commonly associated with proliferation, its context-dependent contributions to epithelial motility are also documented (Cheon et al 2002; Stein et al 2006; Liu et al 2026). Together, we speculate that this combined reduction in junctional rigidity and enhanced β-catenin activity preserves a more dynamically responsive epithelial state, thereby lowering susceptibility to jamming-like phase transitions and limiting challenge-induced arrest.
The functional consequences of this jamming-like state extend beyond impaired tissue remodeling, ultimately compromising periodontal barrier integrity and triggering pathological cascades. In our in vivo model, the failure to reestablish the proper JE attachment apparatus was evidenced by the disorganized spatial distribution of key structural markers (ODAM, Laminin5, and BP180) in WT_P. This barrier defect likely permits the diffusion of bacterial products and damage-associated molecules into the underlying connective tissue, driving robust local immune activation. Consequently, the WT_P group exhibited an exacerbated inflammatory response near the alveolar bone, characterized by elevated IL-1β and IL-6 levels alongside increased Ly6G+ and CD68+ cell infiltration. This localized inflammation occurred concurrently with upregulated RANKL expression and subsequent alveolar bone resorption. These findings support a mechanistic link between the biophysical state of the JE and downstream inflammatory and osteoclastic consequences, providing a cellular explanation for why abnormal epithelial repair can perpetuate bone loss during the early recovery phase.
This biophysical perspective substantially broadens our understanding of periodontal pathogenesis. Classically, P. gingivalis is known to disrupt the epithelial barrier through direct proteolytic degradation of cell adhesion molecules (Hintermann et al 2002; Zheng et al 2021). Our findings introduce a complementary, host-intrinsic pathogenic mechanism: the epithelium mounts a defensive response characterized by junctional reinforcement and reduced motility. However, when sustained into the early healing phase, this response becomes maladaptive, inadvertently arresting the epithelial remodeling required to reestablish cell–tooth attachment. Given that DYNLL1 dysregulation was observed in clinical periodontitis datasets and that Dynll1 haploinsufficiency promoted JE functional reconstruction alongside reduced bone loss in our recovery model, this mechanically altered state likely represents an actionable therapeutic target. Consequently, for early-stage periodontitis and posttreatment management, integrating the modulation of host biophysical properties with conventional antimicrobial strategies may be essential to achieve more predictable and durable periodontal healing outcomes.
We acknowledge several limitations in the current study. First, while the global Dynll1+/− model establishes a clear phenotype, epithelial-specific conditional knockouts are needed to definitively exclude potential contributions from non-epithelial lineages. Second, our single-pathogen model using P. gingivalis does not fully recapitulate the polymicrobial complexity of clinical periodontitis. Third, the current in vivo design did not include ligature-only or P. gingivalis–only groups; therefore, the independent contribution of ligation versus P. gingivalis inoculation to bone loss and JE remodeling cannot be determined from the animal experiments. Finally, although our genetic approach provides robust proof-of-concept evidence, translating these findings into clinical practice requires localized interventions. Future studies will focus on incorporating multispecies biofilm models and developing localized delivery systems (e.g., AAV- or LNP-based vectors) to evaluate the therapeutic potential of targeting epithelial biophysics in vivo.
Author Contributions
Q. Li, contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; J. Li, D. Tao, Y. Che, Q. Yan, contributed to analysis and interpretation, critically revised the manuscript; H. Liu, Y. Zhang, contributed to conception and design, data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261455825 – Supplemental material for Low DYNLL1 Resists Porphyromonas gingivalis–Induced Epithelial Jamming-Like State
Supplemental material, sj-docx-1-jdr-10.1177_00220345261455825 for Low DYNLL1 Resists Porphyromonas gingivalis–Induced Epithelial Jamming-Like State by Q. Li, J. Li, D. Tao, Y. Che, Q. Yan, H. Liu and Y. Zhang in Journal of Dental Research
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Nos. 82220108018 and 82530028 to Y. Zhang; Nos. 82322014 and 82270948 to H. Liu; No. 82370964 to Q. Yan) and the Interdisciplinary Research Project of School of Stomatology Wuhan University (No. XNJC202306 to H. Liu).
Data Availability
All data that support the findings of this study are available from the authors upon request. The raw data for the CRISPR screen and RNA sequencing reported in this article have been deposited in the Genome Sequence Archive (GSA) under accession number HRA016594. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD074035.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.