
Abstract
With the increasing use of fish as model species for research, cell cultures derived from caudal fin explants as well as pre-hatching stage embryos have provided powerful in vitro tools that can complement or serve as an ethically more acceptable alternative to live animal experiments. The widely-used protocols to establish these lines require, as a starting point, homogeneous pools of embryos or viable adult fish which are large enough for collecting sufficient fin tissue. This excludes the use of fish lines with adverse phenotypes or lines that exhibit mortality at early developmental stages and so can only be propagated as heterozygotes. Specifically, when no visually overt mutant phenotype is detectable for identifying homozygous mutants at early embryonic stages, it is then impossible to sort pools of embryos with the same genotypes to generate cell lines from the progeny of a heterozygote in-cross. Here, we describe a simple protocol to generate cell lines on a large scale starting from individual early embryos that can subsequently be genotyped by polymerase chain reaction. This protocol should help to establish fish cell culture models as a routine approach for the functional characterization of genetic changes in fish models such as the zebrafish. Furthermore, it should contribute to a reduction of experiments which are ethically discouraged to avoid pain and distress.
Synopsis
We present a new method for establishing cell cultures from individual zebrafish embryos. As well as contributing new models for experimental research, this protocol will help to avoid the need to propagate fish lines with adverse phenotypes where experiments are ethically discouraged to avoid pain and distress.
Introduction
Fish are well represented amongst the most commonly used model species for research. Notably, the zebrafish (Danio rerio) and medaka (Oryzias latipes) have become popular genetic model organisms for many different biological fields ranging from environmental toxicology, genetics and cancer research to the study of embryonic development. Furthermore, fish cell cultures generated from 24–36 h post-fertilization (hpf) embryos1–6 or from fin biopsies of adult and juvenile fish7,8 have become extremely useful in vitro tools that can complement and serve as alternatives to live animal experiments, particularly in the context of high-throughput analyses. Fish cell lines have been successfully used to screen for drugs, 9 for specific chemical inhibitors and activators of defined biochemical pathways2,10 as well as in large-scale promoter analysis studies.4,11,12 Furthermore, fish-derived cell lines are particularly useful tools in the case of research using protected fish species where the sacrifice of large numbers of fish is prohibitive2,7,13 or in the case of fish lines with adverse phenotypes where experiments are ethically discouraged to avoid pain and distress. Clearly, in vitro experiments cannot fully substitute for in vivo studies; however, the use of fish cell lines at the early stages of a study does enable the development of more focused research questions that can subsequently be addressed at a much more limited scale with whole animal studies.
Current protocols for generating primary cell cultures from early developmental stages of zebrafish required pools of tens to hundreds of embryos in order to have sufficient cells to establish a single cell culture. While this is applicable for homozygous fish lines where all embryos hatching from a clutch of eggs are genetically homogeneous, clearly this is not feasible when dealing with genetically mixed populations of embryos. Mutant lines which exhibit embryonic lethality as homozygotes can be propagated only as heterozygotes. These heterozygotes must be in-crossed in order to generate homozygous mutants in the context of a mixed population which also includes heterozygotes, and wild type progeny. If there is no visually overt mutant phenotype detectable at the 24–36 hpf embryonic stage, then separating pools of sufficient numbers of homozygous mutant embryos for cell line preparation is impossible. In order to resolve this problem, one major technical hurdle to surpass would be to generate and genotype cell lines from individual embryos on a large scale.
Here, we present a simplified protocol to resolve this problem and to obtain homozygous mutant WT as well as heterozygous cell lines starting from mixed pools of 24–36 hpf zebrafish embryos. As proof of principle, here we have used the fbxl3atlv08 mutant zebrafish line and its WT sibling line WTtlv08. 14 This particular mutant was selected due to the lack of morphological distinguishing features of the homozygous mutant embryos and the availability of a robust molecular genotyping method involving polymerase chain reaction (PCR) and subsequent restriction enzyme digestion. After collecting wild type and mutant embryos, each individual embryo is enzymatically dissociated in parallel, then genotyped via PCR and processed to establish an initial ‘pre-cell line’. Then, via a sequential process of pooling together pre-cell lines with identical genotypes and subsequent confirmation of the pool genotype via PCR analysis, it is possible to establish homozygous mutant and wild type cell lines as a source of material for functional comparative studies. A major benefit of this approach is that it avoids the need to eventually raise fish embryos with adverse phenotypes to stages where suffering and distress are unavoidable. This final protocol was established over the course of four trials.
Material, methods and results
Our initial goal was to establish a reliable, rapid and simple technique to generate cell cultures from individual zebrafish embryos at the 24–36 hpf stage. By this approach, we aimed to generate homozygous mutant and WT cell lines from mixed pools of wild type and mutant embryos. More information concerning all the reagents, materials and instruments used for this technique is presented in a detailed list in the Supplementary material file online. We used 24 hpf embryos derived from in-crosses of the zebrafish mutant line fbxl3atlv08 (fbxl3a–/–) together with their WT tlv 08 sibling embryos of the same age (fbxl3a+/+). The fbxl3a–/– line is a CRISPR-generated loss of function mutant for a member of the F-box family, part of the SCF (Skip1/cullin/F-box protein) ubiquitin ligase complex.
Husbandry, breeding and collection of zebrafish eggs
Fish were maintained in a water circulation system at 26°C, fed three times per day with flake food, pellets and living Artemia larvae and maintained under 14:10 h light:dark conditions according to standard methods. 15 The water quality was monitored on a daily basis and maintained at pH 6.8–7.2, with ammonia and nitrite levels of <2 ppm, nitrate levels of <50 ppm and salinity at 300 µS/cm. All husbandry and experimental procedures were performed according to European Legislation for the Protection of Animals used for Scientific Purposes (Directive 2010/63/EU) (General licence for fish maintenance and breeding: Az.: 35-9185.64 Karlsruhe Institute of Technology (KIT)). Regular veterinary inspections (every three months) confirmed that there were no known pathogens affecting the fish in our facility. The fbxl3–/– line 14 is registered in the Zebrafish Model Organism Database (ZFIN) as fbxl3atlv0. Eggs were derived from in-crosses of 6–9-month-old fish. Specifically, for fbxl3a–/– mutants and WT tlv 08 fish, eggs were collected within 3 h following fertilization and washed three times with 1x E3 fish medium (5 mM NaCl; 0.17 mM KCl; 0.33 mM CaCl2; 0.33 mM MgSO4 (Carl Roth)). After removing unfertilized eggs, the eggs were then incubated at 26°C in a petri dish in the presence of 1x E3 medium containing 0.1% methylene blue (Sigma Aldrich) to prevent fungal contamination and maintained under a 14 h light, 10 h dark lighting cycle.
Embryo bleaching
A major challenge during the preparation of cell lines from embryos is to avoid bacterial contamination within the first 72 h following cell dissociation. Zebrafish eggs represent a source of bacteria on the chorion surface. Thus, before cell dissociation, eggs were first bleached in 0.0045% sodium hypochlorite (Carl Roth) using a standard procedure 15 with some modifications (see Figure 1 for a simplified scheme of the starting procedure). In detail, eggs were first transferred into sterile 2 ml safe-lock tubes (Eppendorf) (maximum 25 eggs for each tube) and then washed with D-PBS 1x (Gibco) to completely remove the E3 buffer. Then, eggs were incubated with 0.0045% sodium hypochlorite for 5 min at room temperature in a laboratory test tube rotator mixer (Sarstedt), followed by 5 min incubation in D-PBS 1x, then again by 5 min in the 0.0045% sodium hypochlorite solution followed by 2 × 5 min washing steps with D-PBS 1x.
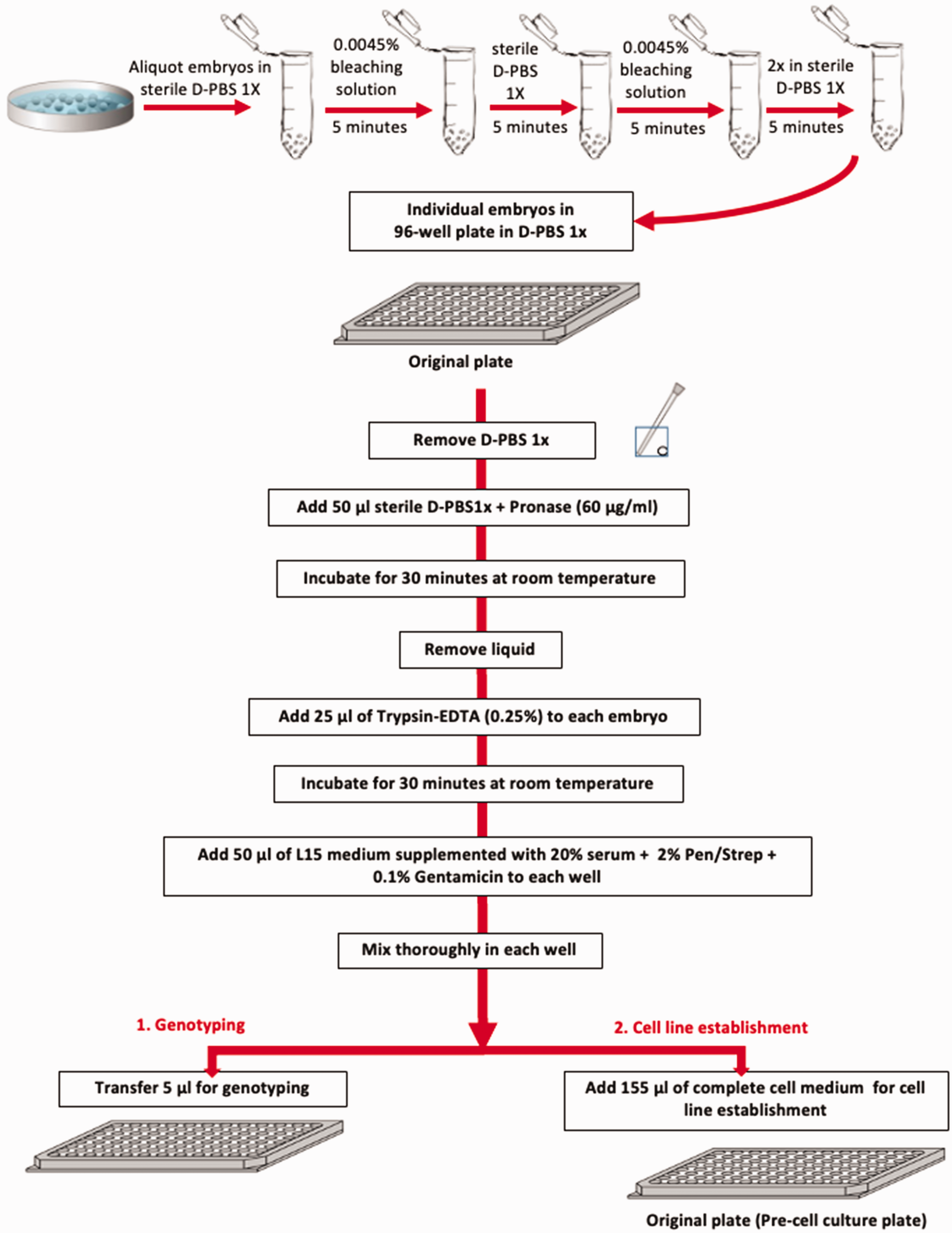
Simplified scheme of the procedure from bleaching of embryos to the beginning of the genotyping and the establishment of the cell lines.
Embryo dissociation
Eggs were then transferred to a biosafety clean bench (Heraeus Deutschland) and individually aliquoted into single wells of a flat-bottomed 96-well plate (Greiner) to facilitate observation using an inverted microscope (DMIL, Leica) during the subsequent procedure (see Figure 1 for a simplified scheme). The transfer of each embryo was performed using plastic Pasteur pipettes (Carl Roth) or P1000 pipette tips (Sarstedt) where the extremities had been cut off to avoid mechanical disruption of the embryos and the risk of cell cross-contamination. From this step onwards, solutions were pipetted into the wells using a multichannel pipette (Integra), again to prevent cross-contamination. Embryos were first treated for circa 30 min to remove or at least soften the protective chorion by adding 50 µl of a 60 µg/ml pronase solution (Sigma-Aldrich) in D-PBS 1x. The concentration of pronase used and the duration of this incubation step were based on our optimized protocol for preparing cell cultures from pools of zebrafish embryos. 1 During this step, embryos were observed through a Stemi 2000 stereomicroscope (Zeiss) to verify the successful weakening or collapse of the chorion. The pronase solution was then removed using a multichannel pipette by tilting the 96-well plate to an angle of circa 30–35°, which allowed a good view of each embryo and thereby avoided the embryos being sucked up inside the pipette tips. Then, the embryos were incubated for 30 min in 25 µl of 0.25% trypsin-EDTA (Gibco) at room temperature to start the cell dissociation process. Dissociation was then blocked by adding to each well 50 µl of Leibovitz’s L-15 complete culture medium (Gibco) supplemented with 20% foetal bovine serum (Gibco), 200 units/ml of penicillin/streptomycin (Gibco) and 0.1 mg/ml of gentamicin (Gibco). These dissociation conditions were based on our optimized protocol for preparing cell cultures from pools of zebrafish embryos. 1 The dissociated embryos were pipetted up and down several times to uniformly distribute the cells/tissue parts inside each well. Visual inspection of the dissociated embryos using the inverted microscope showed that not all the embryos were completely dissociated, with small parts of tails or trunks still visible which seemed to be derived from various parts of the embryo. However, these small trypsin-generated pieces of tissue seemed to be more efficient for subsequently generating healthy cell lines, with fibroblast-like cells migrating out from the tissue pieces (see examples in Figure 2(a) and (b)). At this step, 5 µl of the total cell suspension from each well was transferred to the equivalent position on a 96-well plate (Genomic-DNA plate) already containing 25 µl of freshly prepared 1x lysis buffer solution (10 mM Tris-HCl pH8, 50 mM KCl, 0.3% Tween20 (Carl Roth)) supplemented with 0.1 mg/ml of proteinase K (Thermo Fisher) for genomic DNA extraction (Figures 1 and 3).
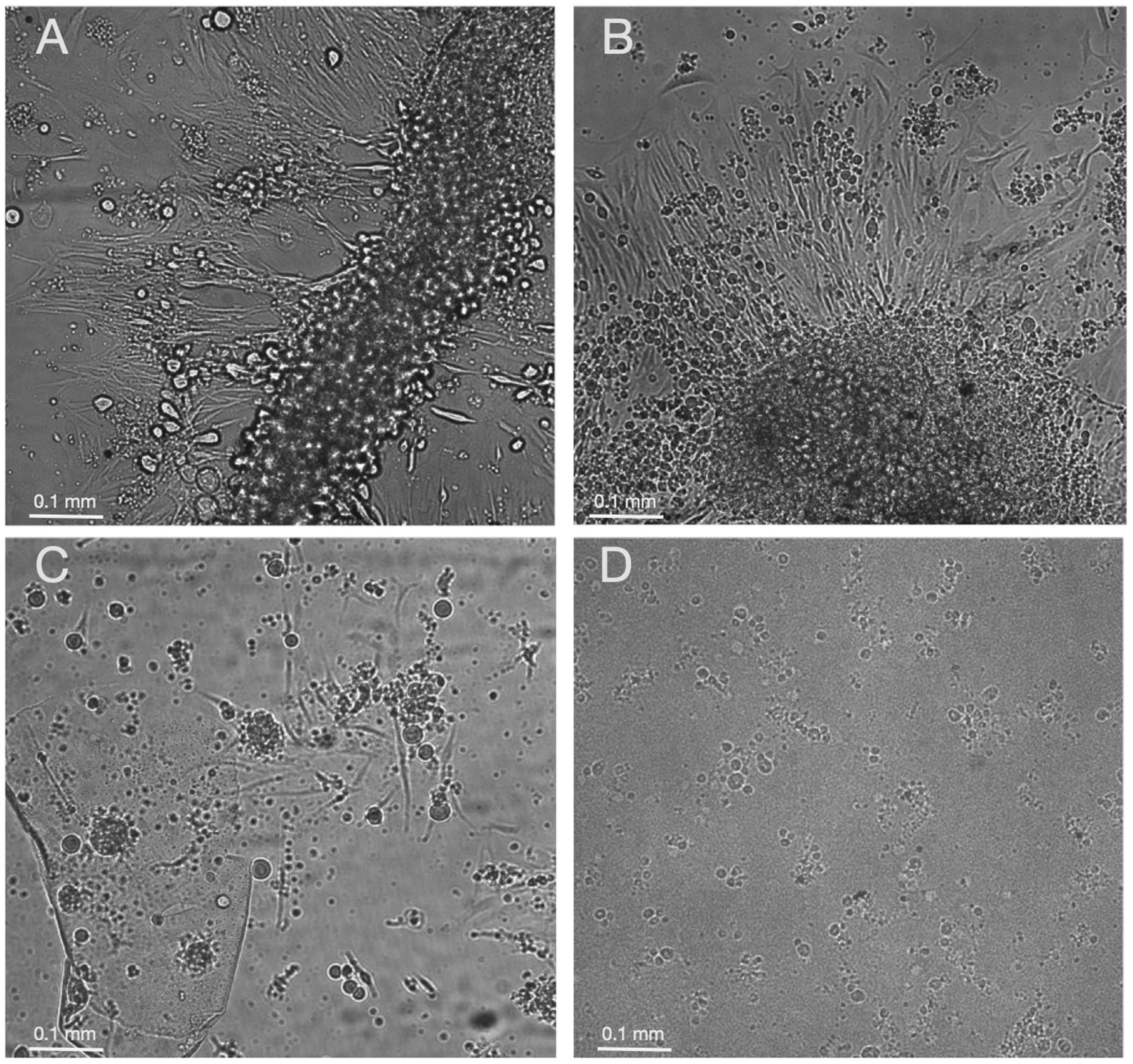
Representative images of dissociated zebrafish embryos after seven days of incubation at 26°C. In panels (a) and (b) fibroblast-like cells are seen migrating out from incompletely dissociated embryo pieces. In panel (c), isolated cells as well as small cell clones derived from fully dissociated embryos are visible. In panel (d), floating, dying cells are visible in the case of an embryo which failed to generate a cell line. Images were acquired using a Leica microscopy system (Dmi8 microscope with a DFC365FX camera and the HC PL FLUOTAR 10x/0.30 DRY objective). A scale bar is included in each image.
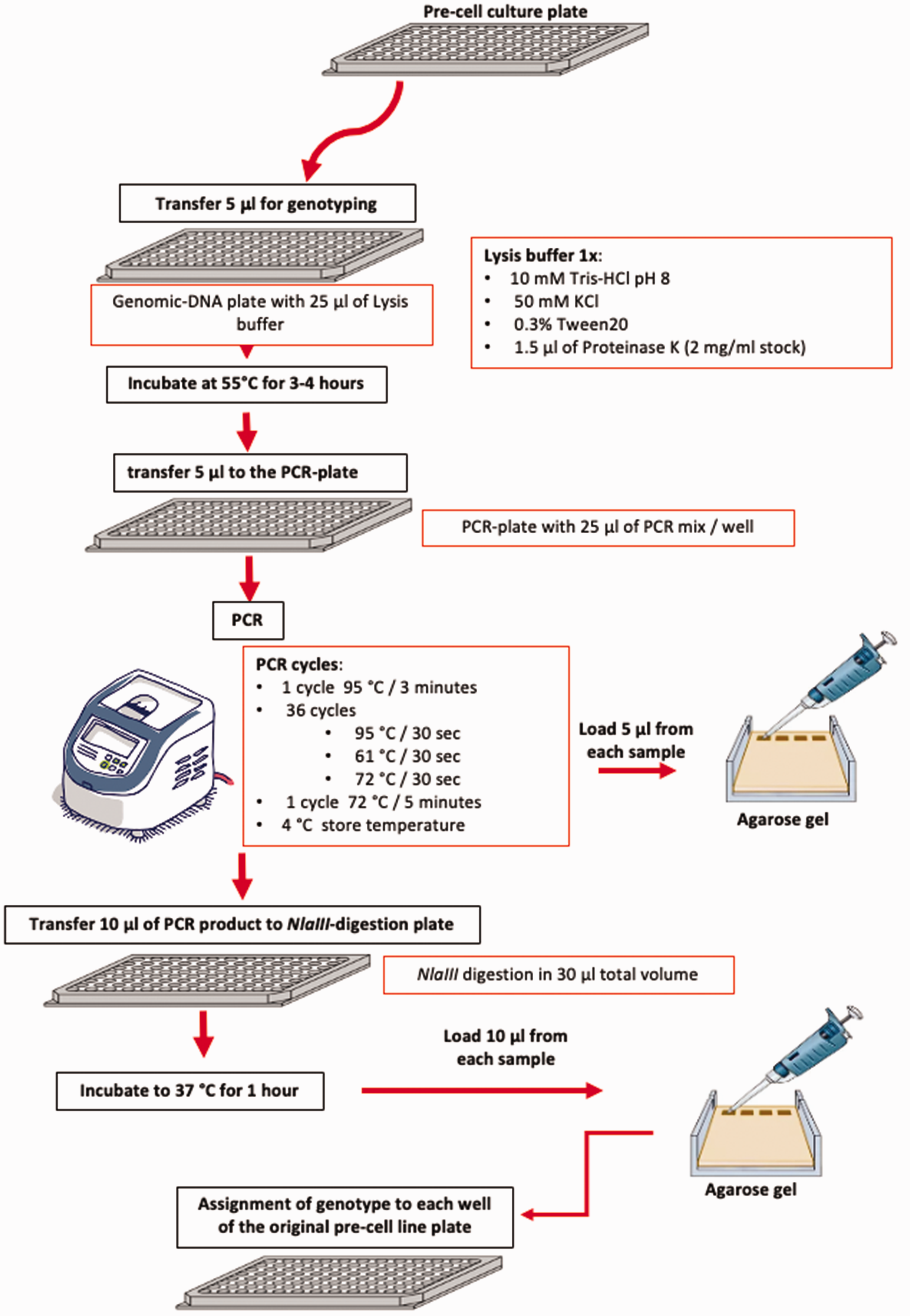
Simplified scheme of the genotyping procedure.
At the same time, 155 µl of L-15 complete medium was added to each well of the original 96-well cell culture plate and the plate was then transferred to an incubator at 26°C (Binder) without supplementary CO2 for several days to allow the cells to attach to the culture substrate, proliferate and to reach confluence. During this process, cell cultures were maintained under complete darkness, apart from the steps when they were inspected or manipulated under normal laboratory lighting conditions.
Genomic DNA extraction and genotyping of zebrafish embryos
The genomic-DNA plate was then incubated at 55°C for 3–4 h to permit genomic DNA extraction followed by inactivation of the proteinase by incubation for 10 min at 95°C and subsequent storage at 4°C until further processing (see simplified scheme of the genotyping procedure in Figure 3).
The genotyping of the 24 hpf embryos was performed via PCR followed by NlaIII restriction enzyme digestion (New England Biolabs) as previously described, 14 with some modifications.
The CRISPR-generated Fbxl3a mutant line (fbxl3a–/–) incorporates a single base pair (bp) deletion within exon 2 which introduces a frame shift and a premature stop codon, resulting in a truncated FBXL3a protein. To distinguish between the different genotypes, we performed a PCR amplification of genomic DNA using primers (Forward: 5′-AGTTGTCACCGAACGAATCTGT-3′, Reverse: 5′-CAAGAAGGGGCAGGTACTGAA-3′ (Sigma Aldrich)), followed by enzymatic digestion with NlaIII that cleaves the DNA sequence 5′-CATG-3′ in the WT allele, but NOT in the mutant allele where the ‘A’ is deleted (Figure 4). To amplify the Fbxl3a sequence, 5 µl of the lysis reaction was pipetted into each well of a new 96-well PCR plate (PCR-plate) (Figure 3). The PCR reaction was performed in a final volume of 30 µl (final concentrations: 1x GoTaq Polymerase Green buffer (Promega), 0.4 µM of the forward Fbxl3a primer, 0.4 µM of the reverse Fbxl3a primer, 0.2 mM dNTPs mix and 5 units of GoTaq Polymerase (Promega). The DNA amplification was performed using a Gene Amp PCR System 9700 (Applied Biosystems) thermocycler set for 3 min at 95°C denaturation followed by 36 PCR cycles of 95°C for 30 s, 61°C for 30 s and 72°C for 30 s. Finally, the samples were incubated at 72°C for 5 min followed by incubation at a storage temperature of 4°C. The PCR products (363 bp for WT and 362 bp for mutant embryos) were visualized by running 5 µl of each reaction on a 2% agarose gel stained with ethidium bromide (Carl Roth) and then examined and documented with the ChemiDoc gel documentation imaging system (Bio-Rad). After visualization of the PCR products, 10 µl from each PCR reaction was transferred into a new 96-well plate (NlaIII-digestion plate) and incubated at 37°C for 1 h with 2.5 units of NlaIII in a final volume of 30 µl. Analysis of 10 µl of the NlaIII-digested PCR products was performed by gel electrophoresis on a 2% agarose gel and then documented with the ChemiDoc imaging system (Figure 5 and Supplementary Figure 1).
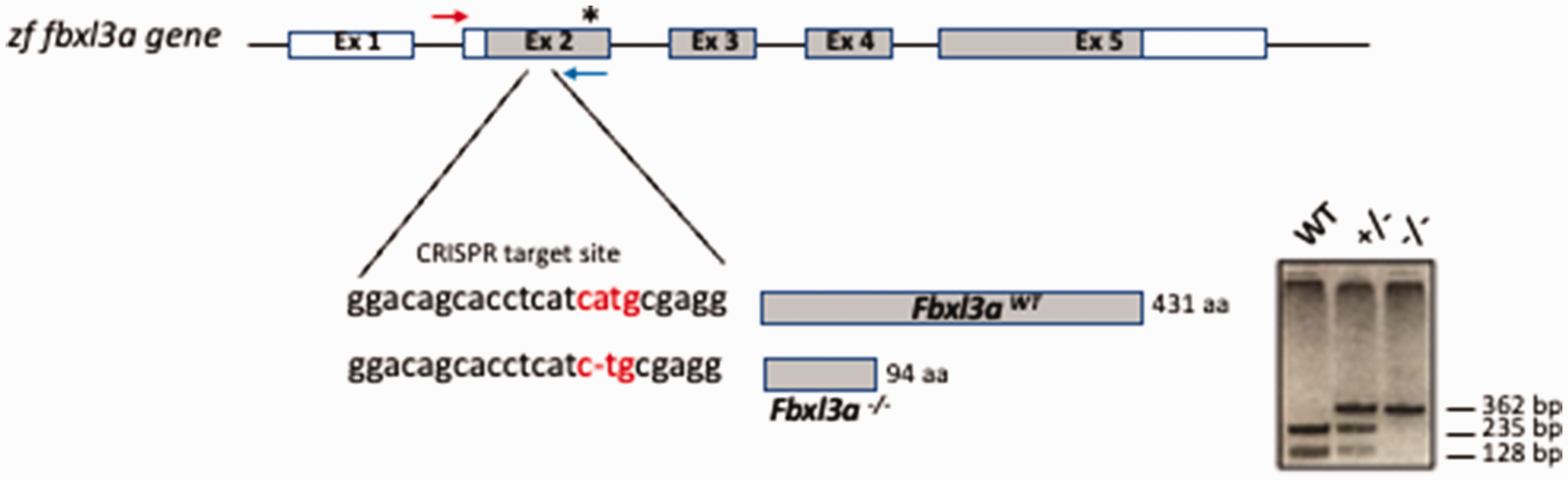
fbxl3a genomic structure and mutation. Genomic organization of the fbxl3a gene. The CRISPR target sequence in exon 2 and the predicted size of the resulting protein (amino acid (aa) length) are depicted. The deletion of a nucleotide A (–) introduces an early stop codon in exon 2 after 80 codons, resulting in a truncated protein. The position of the early stop codon is indicated by an asterisk (
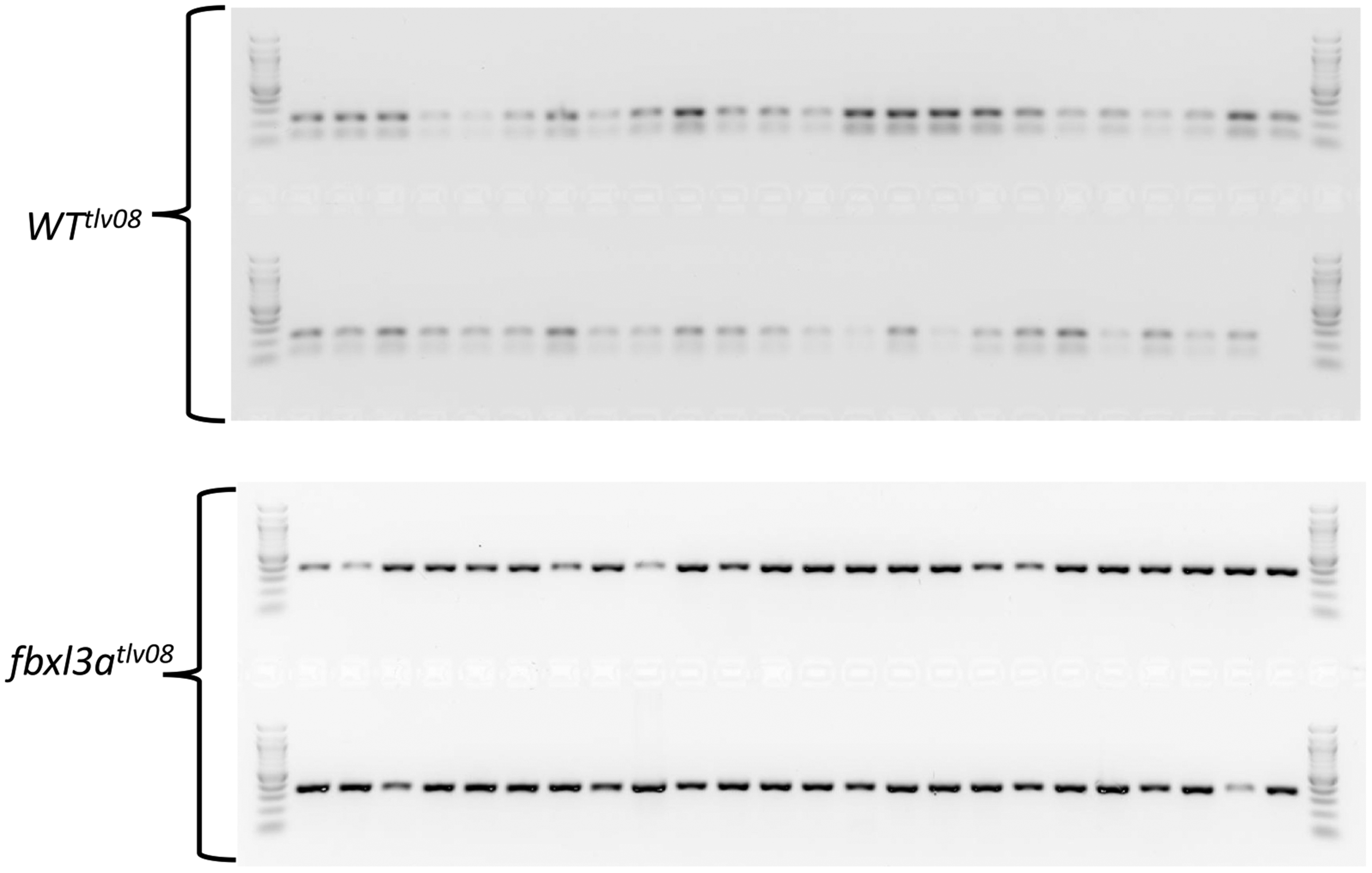
Representative gel of the NlaIII-digested PCR products from WT and fbxl3a mutant embryos. A 100 bp DNA marker (New England Biolabs) is loaded at the beginning and end of each row. Two NlaIII-digested fragments of 235 bp and 128 bp are predicted for the PCR products derived from WT embryos. In contrast, the PCR products from mutant embryos are not digested by NlaIII. Note that the contrast of the image was increased as well as the use of a negative image in order to better visualize the WT tlv 08 samples and the marker wells (see Supplementary Figure S1 online for the original gel images).
PCR products derived from fbxl3a-/- mutant embryos, lacking 1 bp in the NlaIII recognition site, are not digested and so remain as a 362 bp PCR generated fragment, whereas the PCR product generated from WT cells maintains an intact NlaIII site, and so is digested by this enzyme into two fragments of 235 bp and 128 bp (Figures 4 and 5).
Pre-cell culture maintenance
The original 96-well plate with dissociated embryos, which we termed the pre-culture plate, was maintained in an isolated compartment of an incubator at 26°C (without supplemented CO2) to prevent the spread of any bacterial contamination and to allow the cultures to expand without changing the culture medium for circa seven days following embryo dissociation. Wells were checked on a daily basis for possible bacterial or fungal contamination (see Figure 6 for a simplified scheme of the cell line establishment procedure). During the establishment of this protocol, bacterial or fungal contamination developed in 3–6% of the wells with most contamination being observed in the first three to four days. Immediately upon identification of a contaminated pre-culture well, the medium was completely removed, then the well was washed out several times with PBS 1x and dried thereby minimizing the risk of contamination spreading to the adjacent wells. Wells for which the subsequent PCR analysis for genotyping had failed or gave an ambiguous result (circa 5% in total) were also eliminated. Seven days after the start of the culture and onwards, the medium in the pre-culture plate was changed twice per week with fresh complete L-15 culture medium until cell confluence was reached. In approximately 10% of the non-contaminated and successfully genotyped wells, the cell cultures failed to proliferate and were abandoned. Therefore, from a 96-well plate, typically 75–80 confluent wells were obtained from this first step.
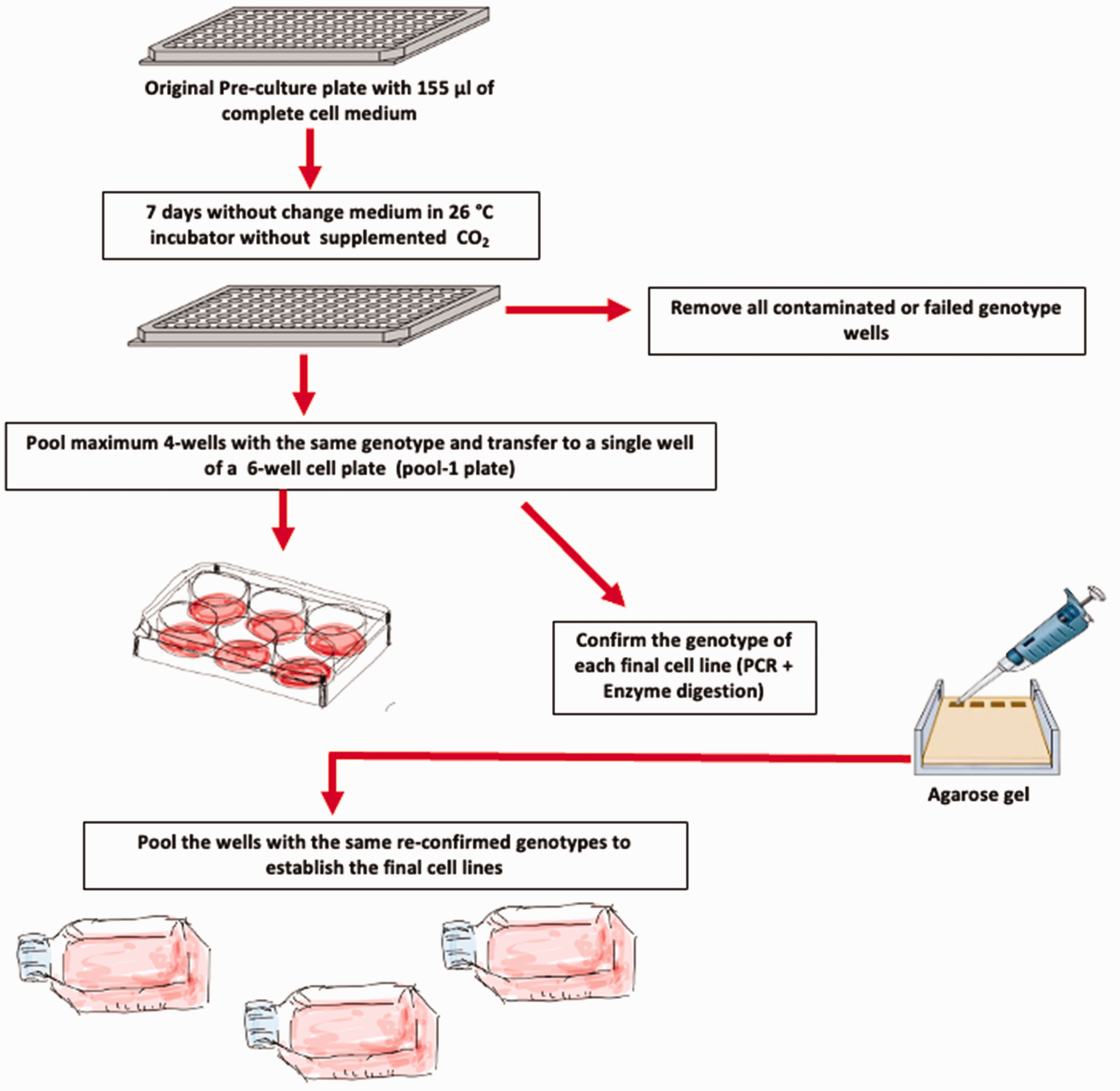
Simplified scheme of the cell line establishment procedure.
Establishment of pooled cell lines
Cell lines originating from individual dissociated embryos of an identical genotype are likely to differ based on which cell types they were originally derived from, as well as genetic changes which arise over the course of the cells adapting to their new, artificial culture environment. For this reason, it is preferable to establish a cell line from a pool of individuals originally sharing the same genotype. In order to avoid incurring any errors during the initial genotyping and culturing process that could ultimately lead to mixed-genotype cell populations in the final cell cultures, we chose not to pool all the putative clones with the same genotype in a single pooling step. Instead, we decided to sequentially pool only three to four lines at a time, which were then genotyped using the same protocol described previously before proceeding to the next pooling step (see scheme in Figure 6). Specifically, at each pooling step, 1/1000 of the total trypsinized cell suspension was used as a source of DNA for the genotyping process.
Discussion
Fish-derived cell cultures have become valuable complementary in vitro models that can be used to address basic questions independently of whole organism complexity. While many protocols have been described for generating fish cell lines, all typically require, as a starting point, pools of genetically equivalent embryos in order to have sufficient numbers of cells to initiate a primary culture.1–6,16 However, in the case of mutants which cannot be raised to sexual maturity as homozygous lines and where genotyping by visual inspection at early embryonic stages is not possible, obtaining homogenous pools of mutant embryos is not feasible. The protocol described here overcomes this limitation by enabling the generation of cell cultures starting from individual embryos and where embryo genotyping can be performed in parallel by PCR analysis of the initial embryo homogenate.
Based on the success rate of this protocol, we predict that it could be readily applied to establish independent cell lines for each possible genotype derived from a single mating between heterozygotes. Specifically, starting from a clutch of circa 200 eggs and considering the possible number of individual embryo lines lost due to bacterial/fungal contamination, genotyping errors or failure of the cells from the embryo homogenate to proliferate sufficiently, our procedure should still ensure that stable cell lines representing all three possible genotypes derived from a single heterozygote in-cross should be readily obtained. Nevertheless, the lethality of certain mutants in the homozygous state may result in fundamental changes to cell biology which could preclude the propagation of these mutant cells in a cell culture environment.
The use of this protocol also represents an important step towards achieving ethical goals such as the 3Rs concept criteria for reducing the size and scale of animal experimentation. It should help to avoid raising fish embryos carrying adverse phenotypes to developmental stages where suffering and distress are unavoidable. This will thereby reduce the need for obtaining experimental authorization for the maintenance of suffering fish at stages in which they can be regarded as independently feeding and free-living animals, within the scope of the European Legislation for the Protection of Animals used for Scientific Purposes. 17
Supplemental Material
sj-pdf-1-lan-10.1177_00236772231157162 - Supplemental material for Establishment of cell lines from individual zebrafish embryos
Supplemental material, sj-pdf-1-lan-10.1177_00236772231157162 for Establishment of cell lines from individual zebrafish embryos by Nathalie Geyer, Sabrina Kaminsky, Shir Confino, Zohar Ben-Moshe Livne, Yoav Gothilf, Nicholas S Foulkes and Daniela Vallone in Laboratory Animals
Footnotes
Acknowledgement
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Author contributions
NSF, YG, DV and SC conceived the study; DV and NSF designed the research project and prepared the manuscript; NG, SK and SC performed the experiments; DV, SC, ZBL, YG and NSF analysed the data; DV and NG prepared the figures.
Data availability statement
The data described in this manuscript are available from the corresponding author, upon reasonable request (
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics statement
All experiments were performed before the embryos’ free-feeding stage and so this study did not require ethical board approval according to the EU Animal Protection Directive 2010/63/EU. General licence for fish maintenance and breeding at Karlsruhe Institute of Technology (DE): Az.: 35-9185.64. The establishment of the fbxl3a mutant zebrafish line used in this study was approved by the Tel-Aviv University Animal Care Committee (04-18-035 and 04-18-051) and conducted in accordance with the National Council for Animal Experimentation, Ministry of Health, Israel.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German-Israeli Foundation for Scientific Research and Development (grant number I-1320-203.13/2015), Helmholtz funding programme NACIP and the Israel Science Foundation (grant number 961/19). We acknowledge support by the Deutsche Forschungsgemeinschaft and the open access publishing fund of Karlsruhe Institute of Technology.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.