
Abstract
Sickle cell disease (SCD) is an autosomal recessive genetic condition characterized by the presence of a mutated form of haemoglobin (HbS). HbS polymerises into long needle-like fibres under low oxygen conditions, leading to the erythrocytes forming sickle shaped red blood cells. With repeated sickling, the red blood cells become irreversibly sickled and trapped within the circulation, and this leads to vaso-occlusive crisis. The patient, a 25-year-old female, previously undiagnosed with SCD, presented with high grade fever, splenomegaly and succumbed due to heat exertion precipitating sickling crisis, multiorgan failure and shock.
Keywords
Introduction
Sickle cell disease is caused by a point mutation in the HBB gene that leads to the substitution of glutamic acid by valine at position 6 of the β-globin subunit of the hemoglobin molecule. 1 The molecules of HbS polymerise to form “tactoids” which are pseudo-crystalline structures formed by deoxygenation. They distort the red blood cells (RBCs) and produce the characteristic sickle shape. Upon reoxygenation, the polymerisation is reversible. However, with repeated sickling cycles the distortion of the cell membrane may become permanent and the RBCs irreversibly sickled. 2
SCD is a multisystem disorder, affecting almost every organ system of the body. The manifestations can be categorized into haematological, vaso-occlusive, infections, and organ dysfunction; 3 with the vaso-occlusive episodes dominating the clinical course. Common clinical features include episodes of ischaemic pain, infarction in spleen, lungs, kidney, liver, bones and central nervous system. 4
Acute multiorgan failure syndrome is a severe, life-threatening complication which presents during a severe pain event, associated with fever, rapid fall in haematocrit and platelet count, non-focal encephalopathy and rhabdomyolysis. 5
This case is an autopsy study that reports on an unusual case of heat exertion that precipitated shock and death in a previously undiagnosed case of sickle cell disease. According to our literature search using the terms “sudden death”, “heat exertion”, “shock” and “sickle cell disease”, there are no articles reporting a case of heat exertion precipitating shock and death in sickle cell disease. Taking into account the fact that this patient with no previous history of illness presented to the hospital with only fever and succumbed to death highlights the importance for pathologists to widen their horizons to consider blood dyscrasias as possible causes for sudden death.
Case history
A 25-year-old woman (a manual labourer) presented with high grade fever of three days’ duration during the peak of the COVID-19 pandemic. She was taken to a primary health centre where she was prescribed paracetamol tablets and then sent back home. The following day, she was brought to the emergency department of the district hospital in Mangalore where she was declared dead. Following which, an autopsy was conducted.
She was moderately built and nourished, weighing 48 kg and measuring 150 cm in length. Both eyes showed yellowish discoloration of the sclera. Internal examination revealed hepatosplenomegaly. The spleen showed multiple foci of yellow-coloured purulent plaque-like material over the diaphragmatic surface. The intima of large vessels in the thorax showed atheromatous streaks.
The spleen weighed 490 gm and the liver weighed 2225 gm which are well above the normal range in women. 6 Hepatosplenomegaly was present. Outer surface of the spleen was bosselated with a few grey-white plaque-like thickenings of the capsule on the diaphragmatic surface. The cut section appeared congested. Grossly, cut surfaces of the lungs, kidneys and liver appeared pale.
Microscopically, the splenic parenchyma showed marked expansion and engorgement of the red pulp with sequestered sickled erythrocytes in the sinusoids (Figure 1 and Figure 2). Additionally, irregular thickening of capsule and trabeculae with multiple haemosiderotic scars were present. The lung parenchyma showed interstitial expansion by fibrosis and inflammatory infiltrate, which was predominantly made up of macrophages. Sequestrated sickled RBCs were seen in the capillaries (Figure 3). Focal osseous metaplasia and megakaryocytes were noted. The renal parenchyma showed congestion and globally sclerosed glomeruli. The proximal convoluted tubules showed features of acute kidney injury, cytoplasmic hemosiderin, luminal hyaline and granular casts (Figure 4). Sequestrated sickled RBCs were seen within the glomerular and interstitial capillaries (Figure 5). Medullary interstitial oedema with patchy fibrosis were also noted. The hepatic parenchyma showed distended sinusoids, packed with sickled erythrocytes (Figure 6), areas of perivenular hepatocellular necrosis, Kupffer cells showing erythrophagocytosis and hemosiderin deposits as well as mild parenchymal cholestasis.
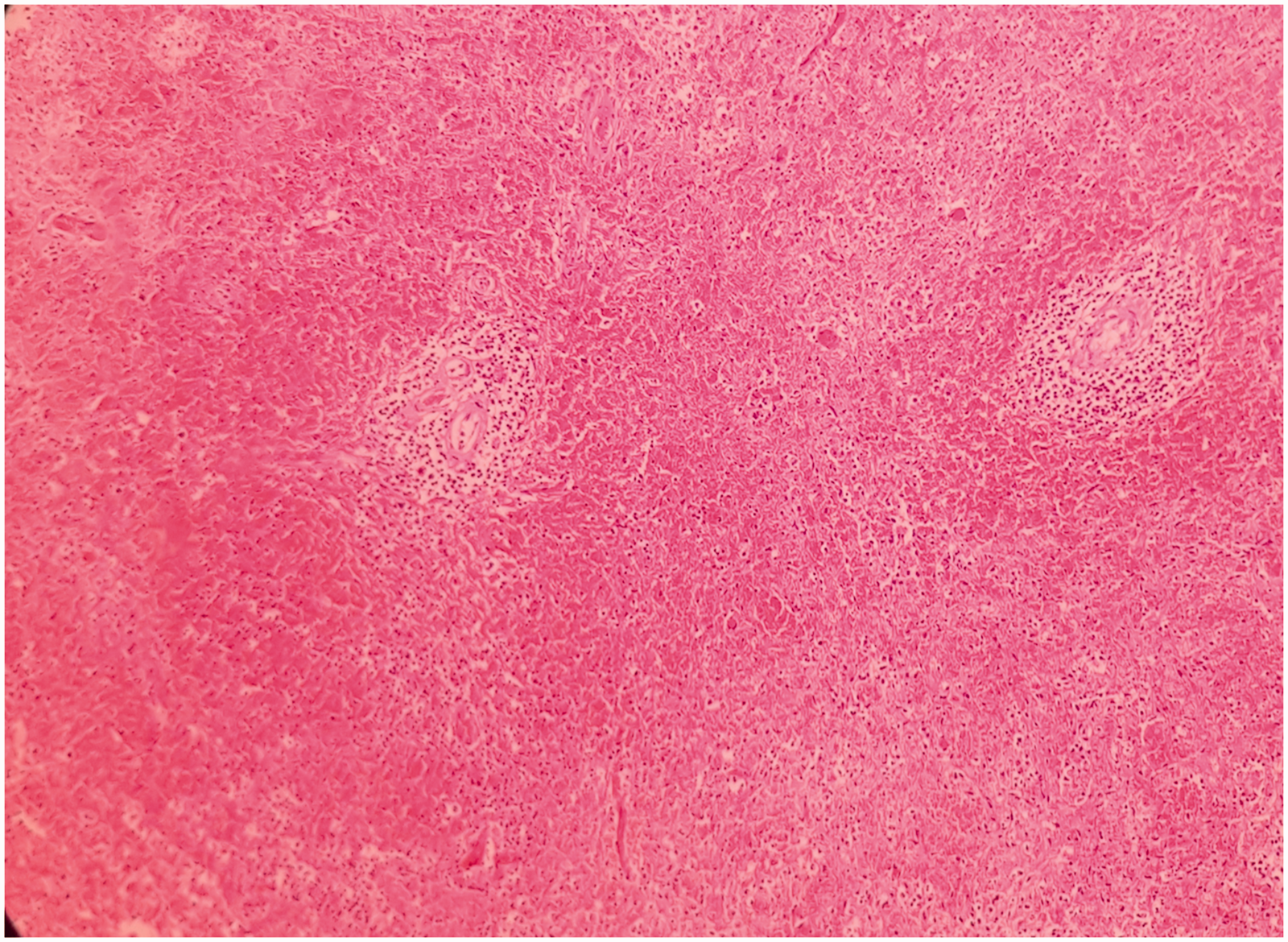
(200x, H&E) – expanded splenic red pulp.
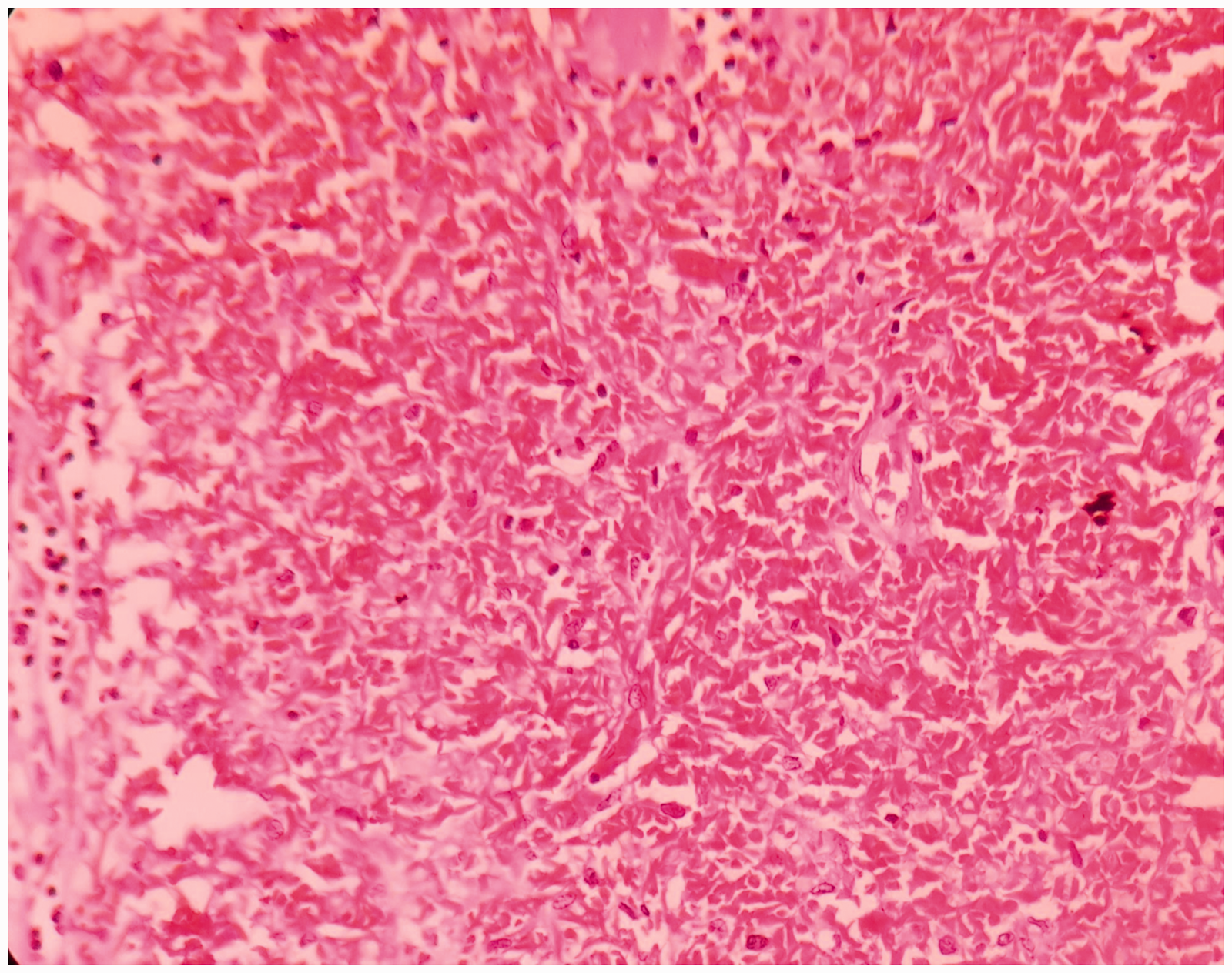
(400x, H&E) – presence of sickled RBCs in the spleen.
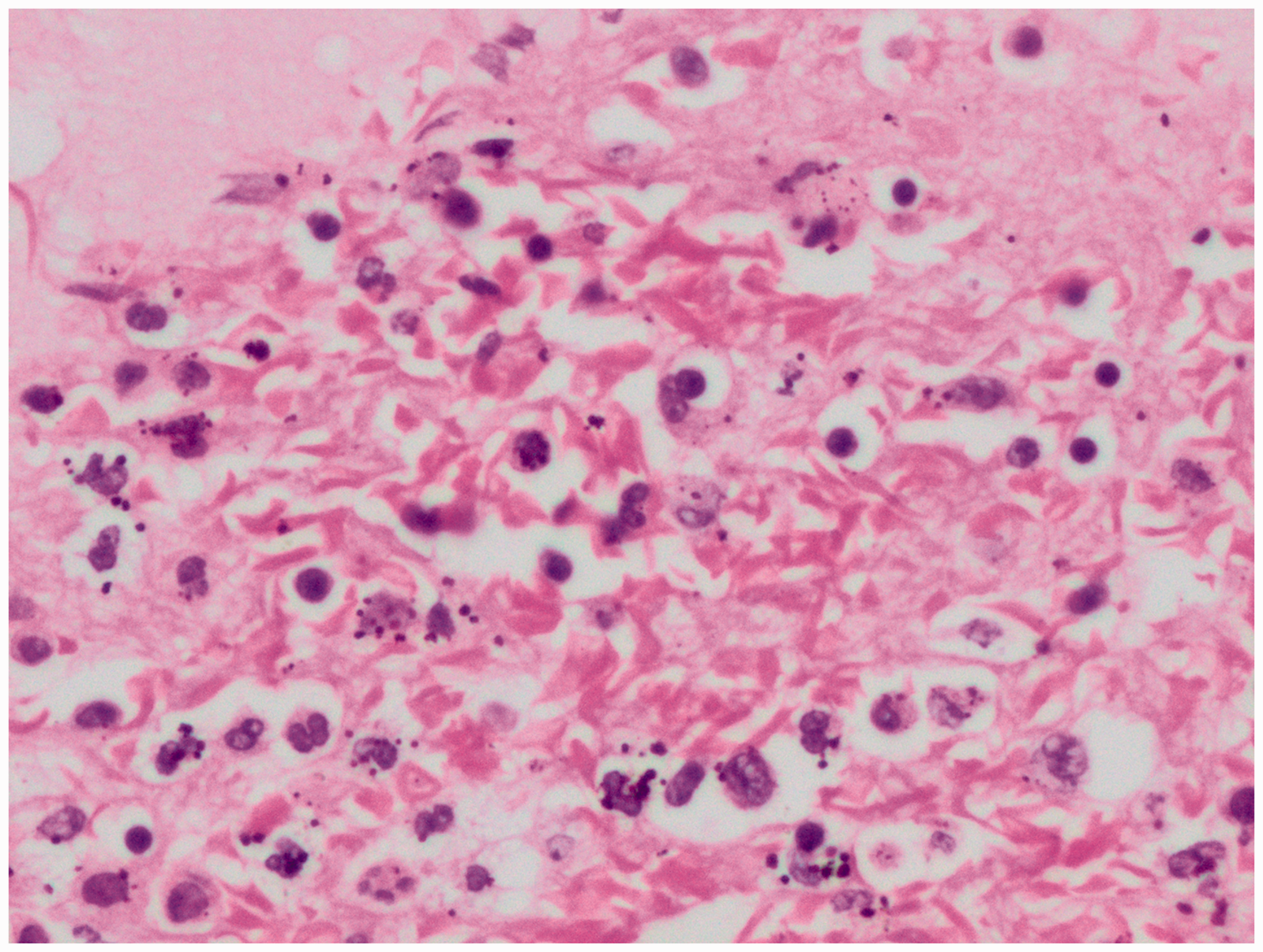
(400x, H&E) – presence of sickled RBCs in the capillaries of the lungs.
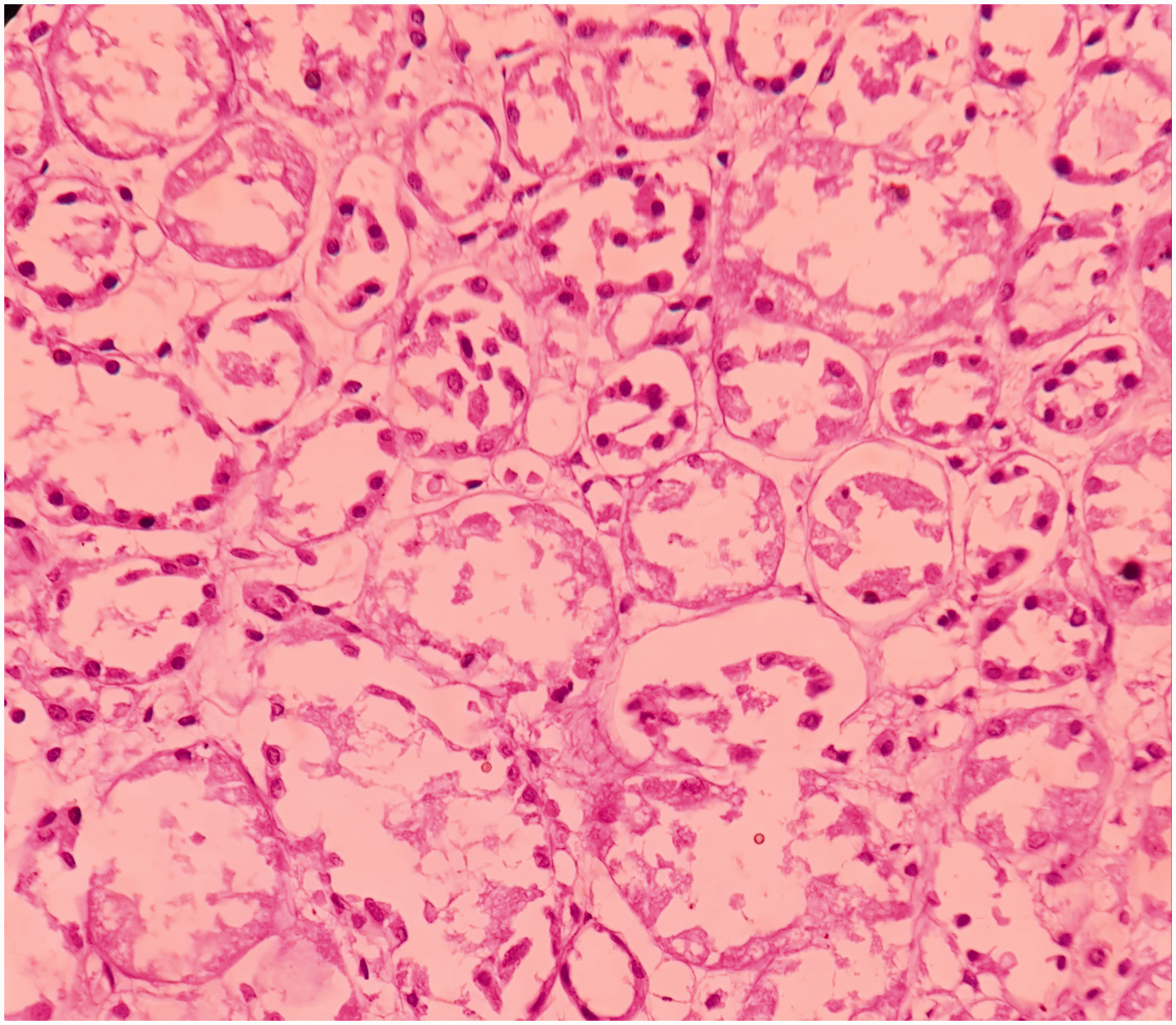
(200X, H&E) – acute tubular necrosis in the kidney.
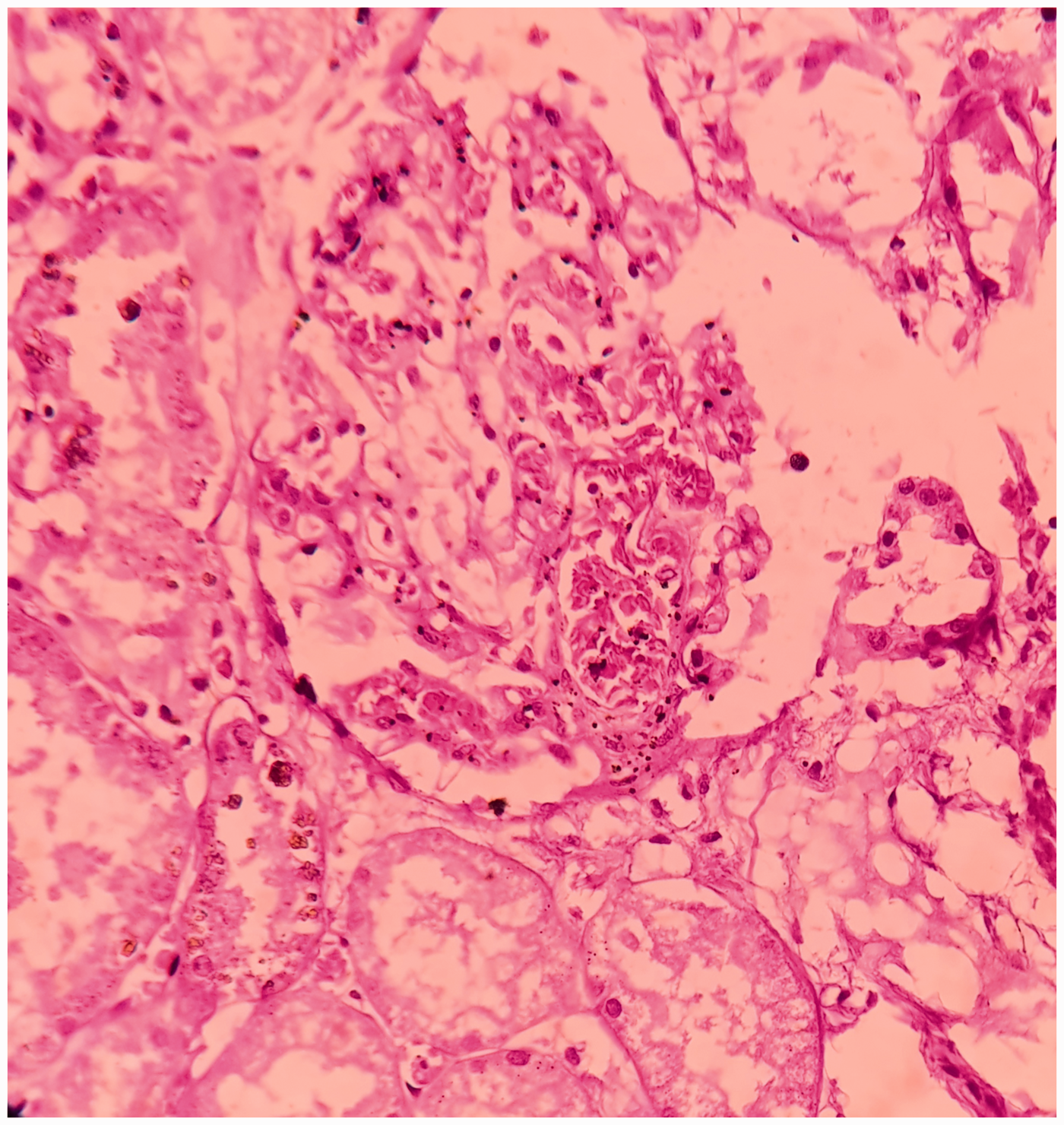
(400X, H&E) – presence of sickled RBCs in the glomeruli.
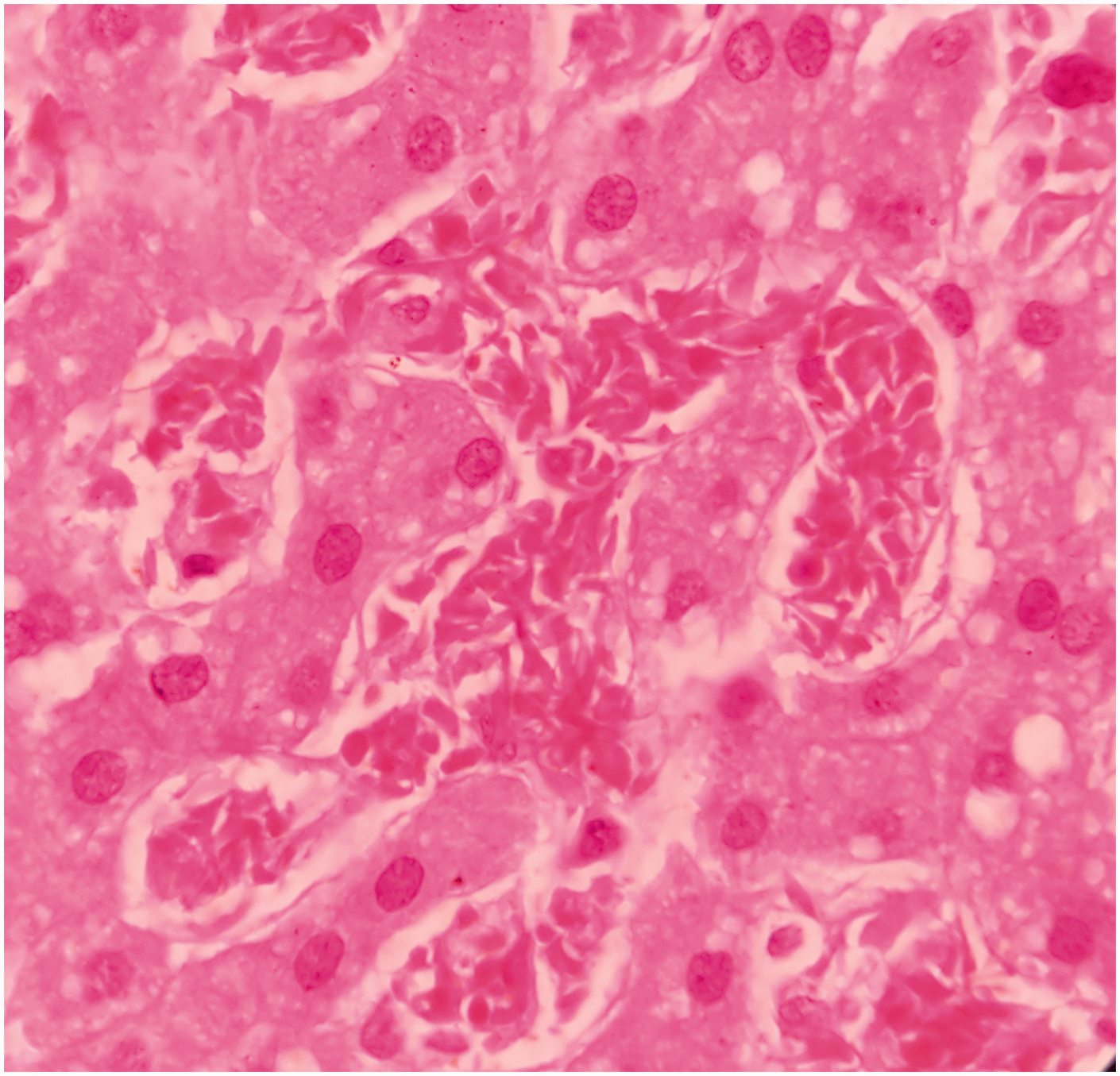
(1000X, H&E) – presence of sickled RBCs in the liver sinusoids.
Discussion
This is an unusual case of previously undiagnosed SCD in a young adult female migrant worker working in Mangalore, a malaria endemic 7 tropical city in southern India. She died from a sickling collapse precipitated by exertional heat illness (EHI). Examination showed hepatosplenomegaly on autopsy. The patient was from the state of Rajasthan, where there is a prevalence of 8.53% SCD and 7.7% sickle cell traits. 8
The three main pathophysiological mechanisms involved in the complications of SCD are vaso-occlusive crisis (VOC), and hyper-hemolytic and infective complications. VOC can present as stroke, priapism, acute chest syndrome, splenic sequestration, liver disease, renal insufficiency etc. Haemolytic complications present as anemia, aplastic crisis, megaloblastic crisis and cholelithiasis. The disease eventually leads to fatal complications like stroke, chronic renal failure, etc. 9
Sickling collapse is an intensity syndrome wherein the sickled RBCs logjam the blood in various organs leading to sequestration syndromes, rhabdomyolysis, shock and multiorgan failure. Triggers include heat, altitude, asthma and over-exertion. Cases of exertional heat illness (EHI) associated sickling collapse in patients with SCT have been reported in varied arenas of life including military training recruits, athletes, sports persons and a medical student who ran a mile in campus. Sickling collapse does not require increased physical activity for prolonged duration. It is described as a “perfect storm” of an overzealous effort for at least a few minutes beyond the fitness level of an individual at that point of time. 10
Though the exact pathophysiology is unknown, Mike et al.11 proposed that there are four pro-sickling factors: local hypoxia, acidosis, increased temperature and RBC dehydration. Exercise can impair tissue oxygen delivery directly or indirectly by local inflammatory response and adhesion molecular expression which can worsen local acidemia and trigger skeletal muscle metabolic failure. 12 In contrast to normal RBCs, sickle cell RBCs show a higher efflux of potassium in response to stress, lower pH and hypoxic conditions that the RBCs encounter during in an exercising muscle. This skeletal muscle metabolic failure along with the potassium efflux leads to catastrophic hyperkalemia causing death. 11
Splenomegaly in this patient was unusual as the spleen becomes atrophic and scarred as explained below. Splenic complications are more commonly found in infants and young children with SCD. At birth the spleen in SCD is normal in size but undergoes a series of changes with age and the replacement of fetal haemoglobin (HbF) with haemoglobin S (HbS). The development of splenomegaly is mainly due to erythrostasis of the sickled RBCs in the anoxic environment leading to marked congestion of splenic pulp and sequestration of erythrocytes. 12 With recurrent vaso-occlusion, infarctions and fibrosis, the spleen regresses in size to a siderofibrotic mass and is no longer palpable and this process is called auto-splenectomy. Most patients beyond the age of 8 years do not have a palpable spleen.
However, splenomegaly can persist in adolescence and adulthood in specific conditions. 12 Patients with sickle cell anemia (SCA) can present with a normal or enlarged spleen into adulthood for the following reasons. In tropical countries low levels of irreversibly sickled cells (ISC) and high levels of HbF reduce the sickling vaso-occlusive events thus maintaining splenic function. In malaria endemic areas chronic antigenic stimulation of the reticuloendothelial system by low parasitic density can result in splenomegaly even in adult SCA patients. Co-inheritance of alpha thalassemia or beta thalassemia with SCA can present with all features of SCA with splenomegaly. 12
Another possibility to be considered is SCT which explains the previously undiagnosed period in this patient and the sudden onset of symptoms following a trigger along with splenomegaly. 9
The presence of environmental factors such as exercise intensity, occupation (manual labourer), tropical climate (heat), dehydration and an area endemic for malaria may have interacted with the genetic profile (HBS) causing the patient's death. 11
The liver sinusoids, glomerular tufts and interstitial vessels of the kidneys and lungs were congested with sickled RBCs with resultant areas of parenchymal and tubular necrosis. Similar histopathological findings were seen in other studies in the literature.13–15
In forensic practice, most cases of sudden death are a result of cardiac pathology. Whereas, in this case, the cause of sudden death was blood dyscrasias, which is rarely suspected or encountered during an autopsy. Darke et al reported that 52.5% of sudden deaths were due to diseases of the heart. 16 It was reported in the USA that most cases of sudden death due to sickle cell disease occur in military personnel or in individuals exposed to heat as a result of their occupation. 17 This case highlights the importance of pathologists considering blood dyscrasias as a cause of sudden death.
Conclusion
In this unusual case of sickle cell trait anaemia, a young migrant labourer, working in high tropical temperatures in a malaria endemic area, developed heat exertion syndrome, precipitating sickling collapse, shock and death. The finding of splenomegaly in a young adult female is unusual which suggests there is a need for more research in this field.
Footnotes
Authors’ contributions
JRP, ACP, JRK and HK were the main contributors to the concept and design of the report, and critically revised the final draft of the article. ACP, MAM and TS collated the case report data from the patient records and drafted the article. All authors critically revised and approved the final manuscript for publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.