
Abstract
Objective
Serum uric acid (SUA) has been associated with cardiovascular and pulmonary vascular disease, but whether SUA has a causal role in pulmonary arterial hypertension (PAH) remains uncertain. This study aimed to evaluate the genetic association between SUA and PAH using Mendelian randomization (MR), with an exploratory population-based analysis in individuals with a higher PAH-related risk-factor burden.
Methods
We conducted two-sample MR using publicly available genome-wide association study (GWAS) summary statistics. The European-ancestry SUA dataset (GCST90014015) was used as the primary exposure dataset, and a cross-population SUA dataset (GCST90018977) was used as exploratory secondary evidence. PAH outcome data were obtained from the largest available PAH GWAS. Cross-sectional NHANES 2003-2018 data were used only as descriptive exploratory context based on a PAH-related risk-factor burden.
Results
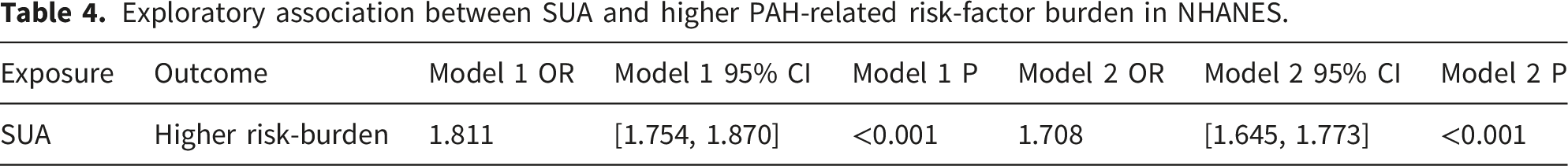
Genetically predicted SUA was associated with higher PAH risk in the European primary analysis (GCST90014015: OR=1.31, 95% CI 1.03-1.68, P=0.028). The mixed-ancestry exploratory analysis showed a consistent direction of effect (GCST90018977: OR=1.52, 95% CI 1.16-2.00, P=0.003), but this estimate was not pooled with the European primary estimate and should not be interpreted as replication because of ancestry mismatch. Sensitivity analyses did not indicate substantial heterogeneity or horizontal pleiotropy. In NHANES, SUA was positively associated with a higher PAH-related risk-factor burden in exploratory logistic models (Model 2: OR=1.708, 95% CI 1.645-1.773, P<0.001).
Conclusion
Our study supports a positive genetic association between SUA and PAH risk. The NHANES analysis provides descriptive exploratory context in a high-risk population. Further research is needed to elucidate the underlying biological mechanisms and confirm these findings in diverse populations.
Keywords
1. Introduction
Pulmonary arterial hypertension (PAH) is a syndrome characterized by progressive pulmonary vascular remodeling, increased pulmonary vascular resistance, and right ventricular dysfunction. According to the 2022 ESC/ERS guidelines, PAH is defined hemodynamically by mean pulmonary arterial pressure >20 mmHg, pulmonary arterial wedge pressure ≤15 mmHg, and pulmonary vascular resistance >2 Wood units measured by right heart catheterization. Because right heart catheterization is required for diagnosis, PAH cannot be directly identified from population surveys without invasive hemodynamic measurements. 1 PAH is a rare disease with a global prevalence estimated to be around 4-14 parts per million derived from a recent systematic review based on epidemiologic data. 2 If left untreated, PAH can progress to right heart failure and death. 3 With the emergence of various targeted therapies in recent years, the 3-year survival rate of some PAH patients has been significantly improved.4,5 However, due to the high cost of treatment, the majority of PAH patients do not receive timely and effective treatment, and the overall mortality rate is still at a high level.6–8 Therefore, finding a biomarker with predictive value for PAH pathogenesis in the early stages of PAH prevention and treatment appears to be urgent.
Serum uric acid (SUA) is the final product of purine metabolism in the human body, mainly excreted through the kidneys. 9 SUA is traditionally believed to be associated with gout and the formation of kidney stones.10,11 However, in the past decade, increasing evidence suggests that elevated uric acid is associated with a variety of cardiovascular diseases and metabolic diseases, such as hypertension, heart failure, diabetes and non-alcoholic fatty liver disease.12–14 Similarly, some observational studies have found an association between uric acid and PAH, but the reliability of the research conclusions remains controversial due to the small sample size and the fact that cross-sectional studies cannot infer the nature of the causal relationship.15,16
Mendelian randomization (MR) is an epidemiological approach that uses genetic variants as instrumental variables. 17 The basic principle of MR is that bases are randomly assembled during meiosis and are therefore essentially random, blind, and minimally biased, greatly reducing confounding factors and reverse causality. It is also considered a small-scale randomized controlled study and is often used to infer the causal relationship between exposure and outcome phenotypes. 18 The present study used two-sample MR as the primary analysis and an exploratory NHANES cross-sectional analysis to examine the association between serum uric acid levels and PAH risk from complementary perspectives.
2. Methods
2.1. Study design
In this study, we first used two-sample MR to explore the causal relationship between SUA and PAH risk. The design of the MR analysis component was strictly in accordance with the guidelines of the Strengthening the Reporting of Observational Studies in Epidemiology by Using Mendelian Randomization (STROBE-MR), and the NHANES cross-sectional component was reported according to the STROBE guidelines.19,20 We provide the comprehensive checklist in Supplementary Material. MR analysis also had to fulfill three assumptions: first, the specific instrumental variables (IVs) used had to be strongly correlated with the exposure phenotype; next, the IVs could not be correlated with confounders; and lastly, the IVs could only influence the outcome phenotype through the exposure phenotype (Figure 1). Diagram of the core assumptions of mendelian randomization.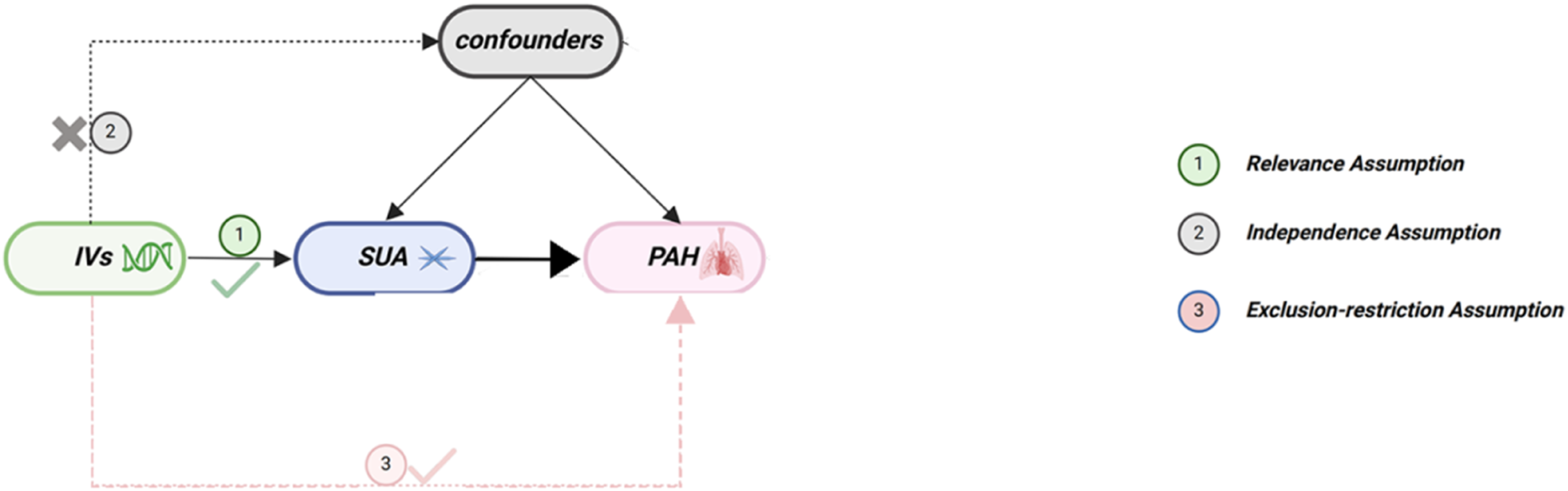
On the basis of TSMR analysis, we additionally used NHANES 2003-2018 data to provide exploratory population-level context for the genetic findings. Because GCST90018977 was a mixed-ancestry SUA genome-wide association study (GWAS) whereas the PAH outcome GWAS was European, estimates from the European primary SUA dataset and the mixed-ancestry exploratory SUA dataset were reported separately (Figure 2). Flowchart of the revised study design including mendelian randomization and exploratory NHANES analyses. The European primary MR estimate and mixed-ancestry exploratory estimate were reported separately because of ancestry mismatch. NHANES was used to evaluate a PAH-related risk-factor burden.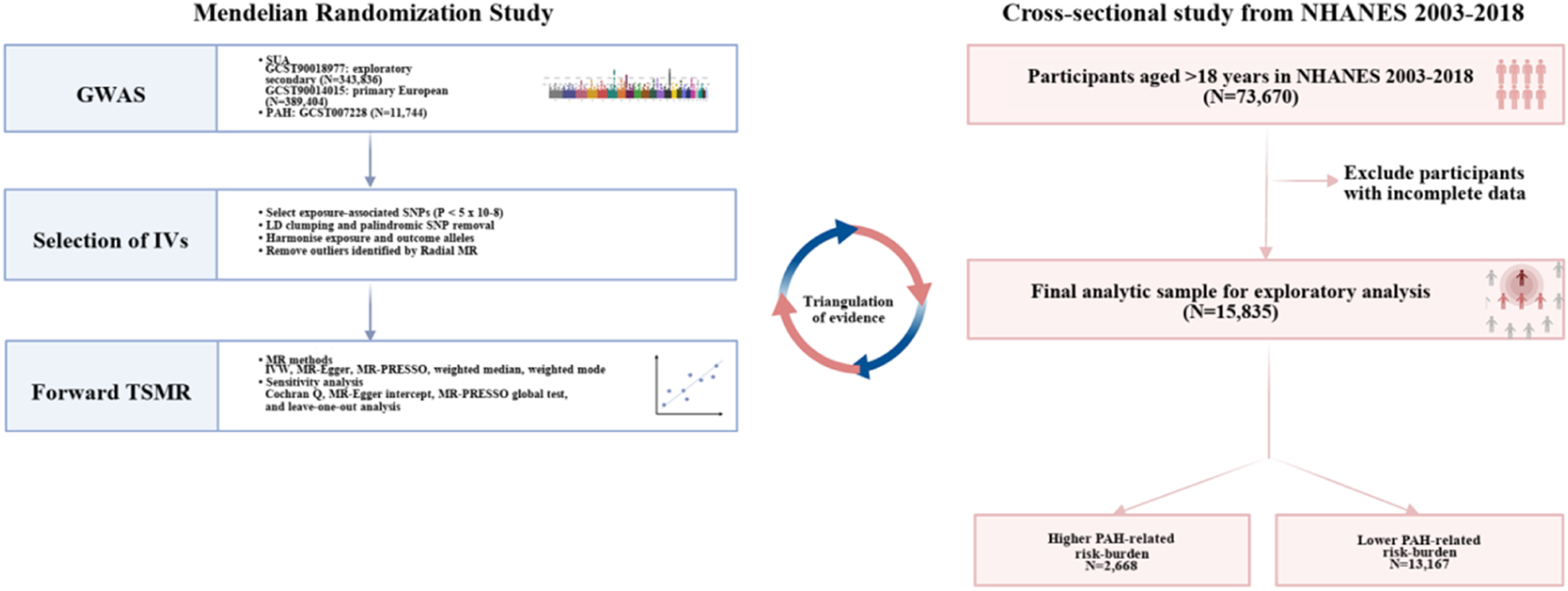
2.2. Data sources
Details of the GWAS datasets used in the revised MR study.
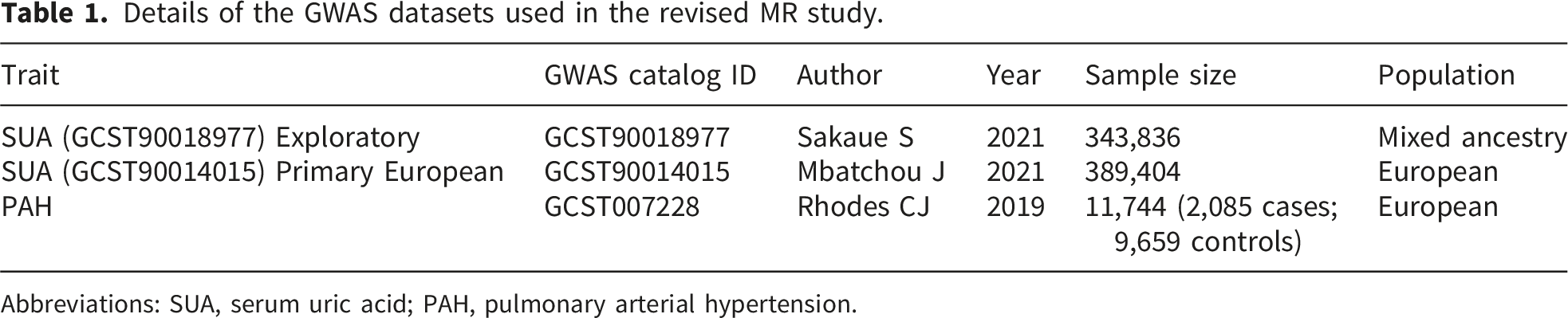
Abbreviations: SUA, serum uric acid; PAH, pulmonary arterial hypertension.
Cross-sectional data were obtained from NHANES 2003-2018. These cycles were selected because they were the most recent standard continuous 2-year NHANES cycles available for the prespecified harmonized analysis with consistent SUA and covariate data; later releases would require separate harmonization because of the special 2019-March 2020 pre-pandemic files and changes in the 2021-2023 cycle. Because NHANES does not include right heart catheterization or a verified PAH diagnosis, we did not define NHANES participants as PAH cases. Instead, we constructed an exploratory PAH-related risk-factor burden using available variables related to pulmonary vascular and cardiometabolic risk. The exploratory PAH-related risk-factor burden was adapted from prior NHANES work on PAH high-risk populations. Because SUA was the exposure of interest in the present study, SUA itself was not used to define the risk-burden phenotype. In the harmonized analytic dataset, the available non-SUA components included systemic hypertension, diabetes mellitus, older age, obesity, sex, and blood pressure-related measures. Participants meeting the prespecified combined risk-factor profile were classified as having a higher PAH-related risk-factor burden. This analysis was intended only to examine whether SUA tracked with a clinically relevant high-risk phenotype in the general population, and variables with missing values in the subset data were excluded. Other covariates included in the article were SUA, age, sex, diabetes mellitus (DM), hypertension, body mass index (BMI), total bilirubin (TB), systolic blood pressure (SBP), and diastolic blood pressure (DBP), hemoglobin (HB), white blood cell (WBC), red blood cell (RBC), monocyte, mean platelet volume and blood platelet count (PLTC). In the final data analysis, participants without any PAH risk factors other than the PAH group were included in the control group, resulting in a total of 15,835 patients, including 2,668 patients in the PAH group and 13,167 in the control group, as detailed in Supplementary Table 3. All GWAS and clinical data used in this study were obtained from published studies or public databases, and subject consent was obtained and reviewed by the ethics committee in the original study.
2.3. Statistical analysis
We used single nucleotide polymorphisms (SNPs) that were strongly associated with the exposure phenotype (p < 5 × 10-8) as IVs, while setting R2 = 0.001 and window size = 10,000 kb as the thresholds for screening independent SNPs. For TSMR analysis, we adopted the inverse variance weighted (IVW) method as the primary MR analysis method, which obtains the final causality estimate by calculating the weighted sum of effect estimates for all SNPs. However, considering that IVW ignores null IVs and heterogeneity, we additionally supplemented other MR methods to increase the reliability of the results, including MR-Egger, Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO), Weighted median and Weighted mode.
For the analysis of baseline data, we utilized the Mann-Whitney U test for continuous variables (presented as median and interquartile range [IQR]), and the chi-squared test for categorical variables (presented as frequency and percentage [%]). Furthermore, we explored the association between SUA levels and the higher PAH-related risk-factor burden using univariate and multivariate logistic regression analyses. The results are presented as OR with 95% confidence intervals (95% CI). Model 1 results are based on univariate logistic regression without any variable adjustments. Model 2 adjusts for sex, age, and BMI on the basis of Model 1. All analyses in this study were performed in the R language environment (version 4.3.1), and two-sided p-values less than 0.05 were considered statistically significant.
2.4. MR sensitivity analyses
To evaluate the stability of the MR findings, we performed several sensitivity analyses. Cochrane’s Q-test was used to assess heterogeneity, and the ‘leave-one-out’ method to explore the possibility of excluding individual SNPs altering the overall causality. In addition, we performed MR-Egger intercept test, Radial MR to test for outliers and horizontal pleiotropy. All analyses related to TSMR were performed using the “TwoSampleMR”, “MRPRESSO” and “RadialMR” R packages.
3. Results
3.1. MR analysis
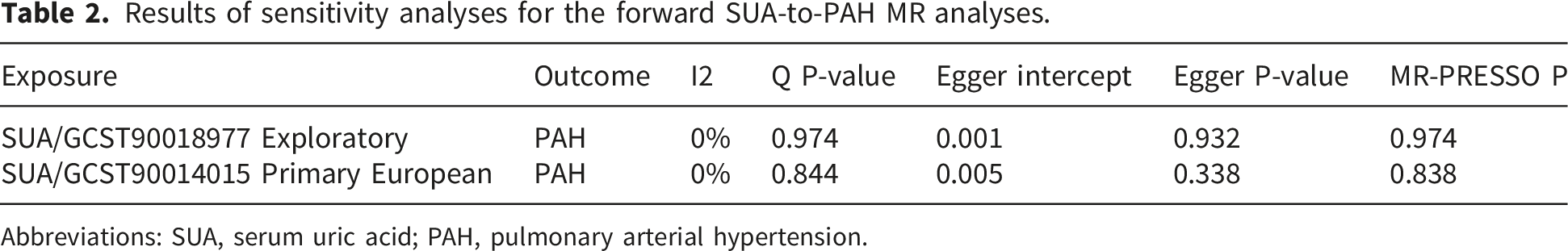
We implemented stringent SNPs screening criteria to ensure that the IVs in the analysis were strongly associated with exposure and not associated with outcome versus confounders. We ultimately included 226 and 237 SNPs from the GWAS datasets of the two SUAs for MR analysis, respectively, and all SNPs had F-values well above 10, which indicated the absence of weak instrumental variable bias, and SNP-specific information is detailed in Supplementary Tables 1. The outlier SNPs identified and excluded by radial MR are displayed in Supplementary Table 2. Based on the IVW approach, our results showed that there was a significant positive causal association between SUA levels and PAH (Supplementary Figure 1), populations with high SUA levels have genetically causally higher risk of developing PAH (Figure 3). In the discovery cohort, genetically predicted SUA was associated with a higher risk of PAH (OR=1.31, 95% CI 1.03-1.68, P=0.028). The exploratory dataset showed a consistent positive association (OR=1.52, 95% CI 1.16-2.00, P=0.003). Given the ancestry mismatch in GCST90018977, this estimate was reported separately and used only to assess directional consistency; it was not pooled with the European primary estimate and should not be interpreted as formal replication or as a combined causal estimate. In subsequent sensitivity analyses, Cochrane’s Q-test, MR-Egger Intercept test and MR-PRESSO results showed no heterogeneity in both discovery cohort and validation cohort results (Table 2). In both the “leave-one-out” and radial MR analyses, the results did not exhibit the presence of anomalous outliers and horizontal pleiotropy, suggesting that our results are robust to heterogeneity and pleiotropy (Supplementary Figures 2 and 3). Forest plot of the association between genetically predicted SUA and PAH. GCST90014015 was used as the European primary SUA dataset, and GCST90018977 was used as mixed-ancestry exploratory secondary evidence. Higher PAH-related risk-factor burden was defined using a prespecified combined profile of available non-SUA PAH-related risk factors. SUA was not used to define this phenotype. This exploratory phenotype did not represent clinically diagnosed PAH. Results of sensitivity analyses for the forward SUA-to-PAH MR analyses. Abbreviations: SUA, serum uric acid; PAH, pulmonary arterial hypertension.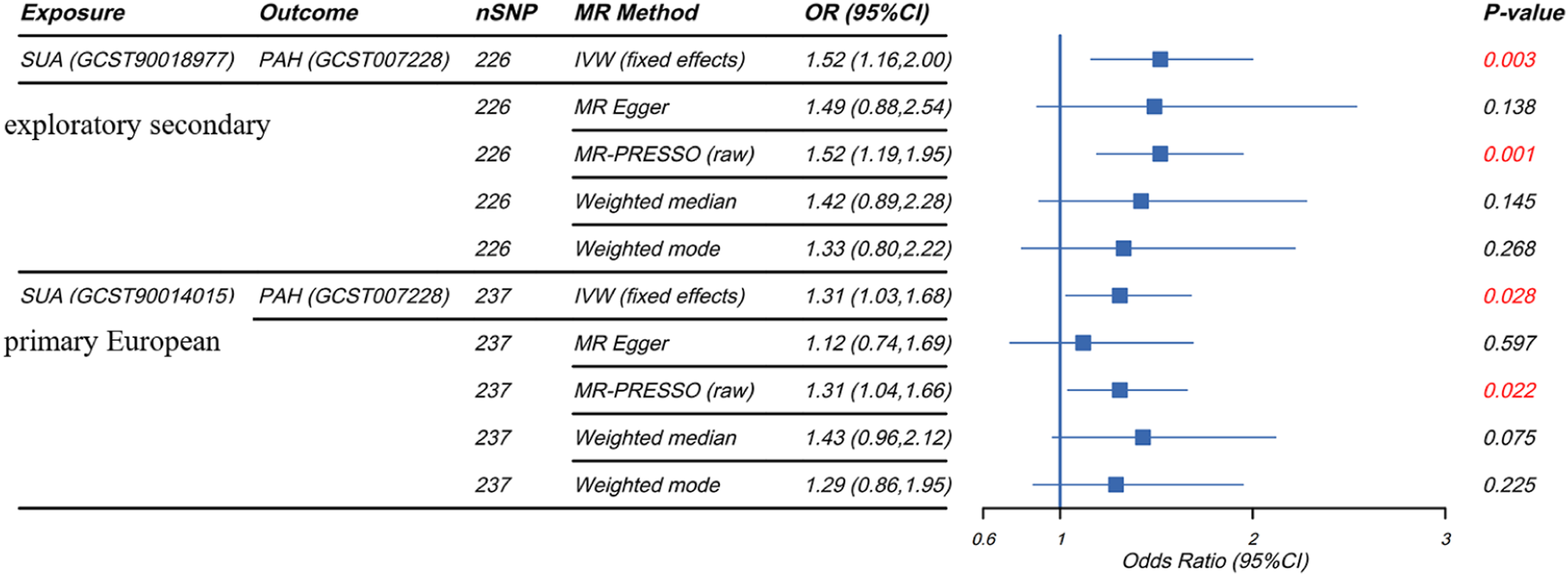
3.2. Exploratory analysis in NHANES
Baseline characteristics of NHANES participants stratified by PAH-related risk-factor burden.
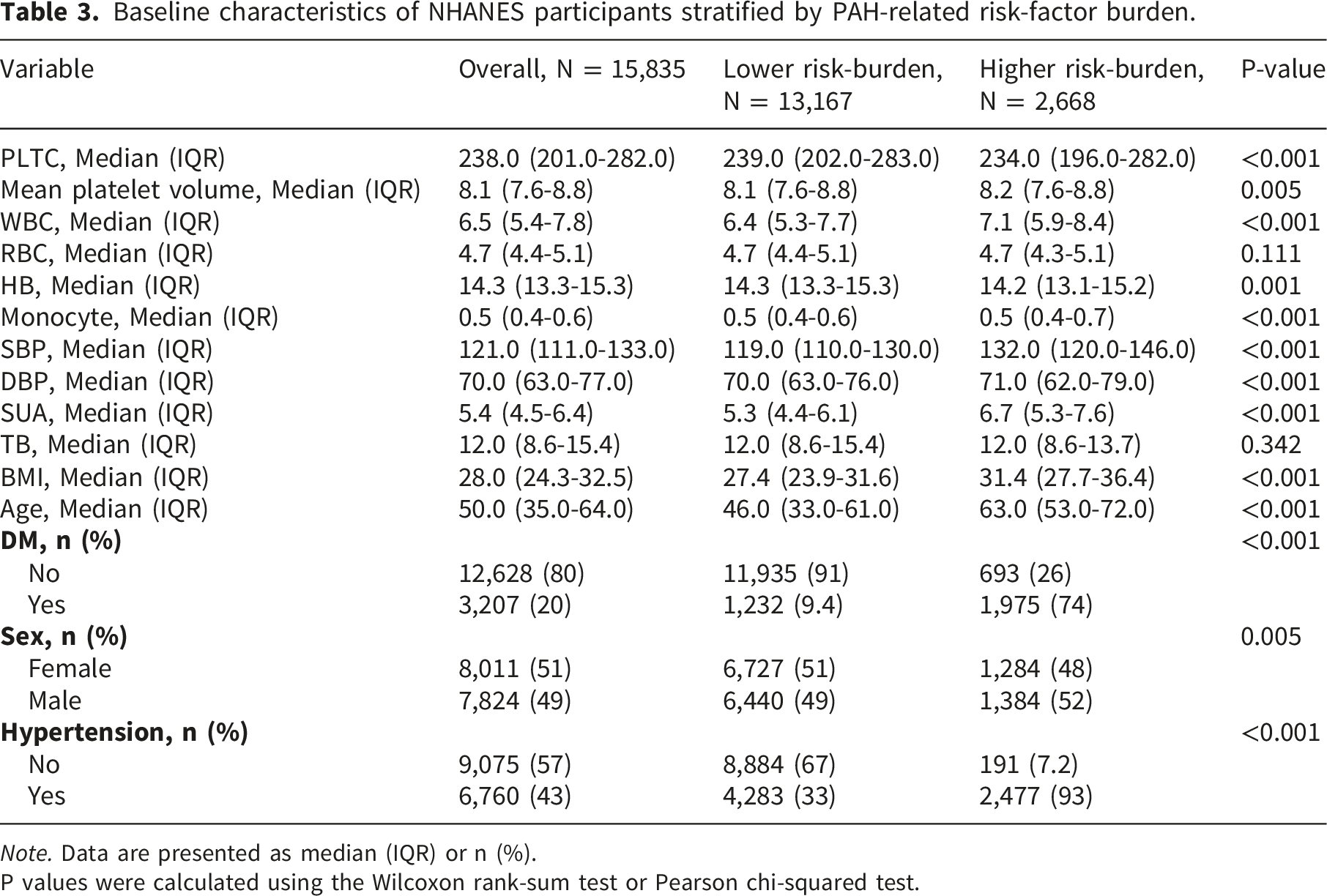
Note. Data are presented as median (IQR) or n (%).
P values were calculated using the Wilcoxon rank-sum test or Pearson chi-squared test.
Exploratory association between SUA and higher PAH-related risk-factor burden in NHANES.
4. Discussion
In this study, in the primary MR analysis conducted in a European population, genetically predicted SUA levels were positively associated with the risk of PAH; furthermore, a GWAS of SUA in a population of mixed ancestry, serving as exploratory secondary evidence, also demonstrated effects consistent in direction. The NHANES analysis was regarded solely as an exploratory analysis concerning the burden of risk factors associated with PAH, rather than as a validation or replication of the MR study findings. Overall, the results of this study support a positive genetic association between SUA and the risk of PAH.
The clinical association between SUA and pulmonary vascular disease has been reported for more than two decades. Early work showed that higher SUA was associated with worse hemodynamic severity and mortality in primary pulmonary hypertension. 24 Subsequent studies in connective tissue disease-related PAH and systemic sclerosis-related PAH also found that higher SUA was associated with higher pulmonary vascular resistance, poorer functional status, and worse clinical outcomes.15,16 In addition to PPAH, SUA demonstrated strong clinical value in secondary PAH. A cross-sectional study found that in patients with systemic lupus erythematosus, SUA was significantly higher among patients with PAH than among these with normal PAP. 25 Taken together, these findings support SUA as a clinically relevant pulmonary vascular marker, while observational associations should still be interpreted cautiously. In addition, interpretation of clinical and genetic evidence depends on accurate PAH phenotyping. PAH should be distinguished from pulmonary hypertension due to left heart disease and other pulmonary hypertension subtypes through standardized diagnostic evaluation and right heart catheterization. 26 Recent studies also suggest that right ventricular phenotyping, including right ventricular remodeling and right ventricular-pulmonary arterial coupling, may improve disease characterization and risk assessment in PAH.27,28 Future PAH GWAS datasets with hemodynamically confirmed diagnoses, detailed subtype classification, and deeper cardiopulmonary phenotyping would further strengthen genetic studies of PAH.
Several biological pathways may explain the positive association between higher SUA and PAH risk, including endothelial dysfunction, impaired nitric oxide signaling, inflammation, oxidative stress, altered purine metabolism, and pulmonary vascular remodeling.29–31 These mechanisms are biologically plausible and supported by experimental or translational studies. Endothelial dysfunction is central to PAH pathobiology. Dysfunctional pulmonary arterial endothelial cells contribute to vasoconstriction, inflammation, thrombosis, and vascular remodeling through imbalance of nitric oxide, endothelin, prostacyclin, and inflammatory signaling. 29 SUA may aggravate this process by reducing nitric oxide bioavailability and impairing endothelial homeostasis. 32 Experimental evidence suggests that increased lung uric acid can enhance arginase activity, decrease cGMP signaling, and worsen pulmonary vascular responses in PAH models. 31 In addition, SUA can bind to the transcription factor activator protein-1 binding site to induce endothelin-1 (ET-1) gene expression. 33 ET-1 can induce an increase of the expression of lysyl oxidase in pulmonary artery smooth muscle cells, causing ectopic deposition of elastin and an increasing pressure in pulmonary artery. 34 These findings provide a plausible link between elevated SUA and abnormal pulmonary vasoconstriction, although translation to human PAH still requires further validation. Altered purine metabolism may also connect SUA biology with pulmonary vascular remodeling. PAH is increasingly recognized as a disease involving metabolic reprogramming of pulmonary vascular cells. 30 Proliferating pulmonary artery smooth muscle cells require purine nucleotides for DNA and RNA synthesis, and recent experimental work showed that suppression of de novo purine synthesis attenuated pulmonary vascular remodeling and pulmonary hypertension in rodent models. 35 Because SUA is the final product of purine metabolism in humans, these data support the broader biological relevance of purine metabolic pathways in PAH, even though serum SUA itself may reflect only part of this metabolic network.
The renin-angiotensin-aldosterone system (RAAS) is an important hormone system in the body that regulates blood pressure and fluid balance. 36 In the rat model experiment, high levels of SUA, rapidly taken up by vascular smooth muscle cells (VSMCs), may upregulate the expression of angiotensinogen messenger RNA in the cells, leading to the production of angiotensin II(AngII), resulting in RAAS activation and VSMCs proliferation. 37 AngII, the most potent vasoconstrictor known, produces a biological effect by binding to angiotensin II type I receptors on vascular endothelial cells, 38 which induces constriction of small arteries throughout the body including the pulmonary arteries, resulting in elevated pulmonary arterial blood pressure ultimately progressing to PAH.39,40 In addition, Ang IIcan stimulate the secretion of aldosterone in the adrenal cortex to induce pulmonary artery vascular remodeling, hypertrophy of VSMCs and increase the reabsorption of water and sodium in the body, further rising PAP.41,42
Immune inflammatory factors play an important role in PAH formation and vascular remodeling processes.43,44 Some studies have shown that SUA can stimulate endothelial cells to release various pro-inflammatory molecules, which can participate in the inflammatory response and lead to the proliferation of vascular smooth muscle cells.45–47 SUA increases the gene expression of inflammatory cytokines, including interleukin-6 (IL-6) and interleukin-8, tumor necrosis factor-alpha, and miR-155, by activating nuclear factor kappa B.48,49 The serum concentration of IL-6 is significantly elevated in patients with severe primary PAH. 50 A multicenter study in pediatric patients with PAH has shown that elevated serum IL-6 is significantly associated with higher PVR index, greater mean PAP, and worse clinical prognosis. 51 Higher concentration SUA stimulates upregulation of miR-155 expression in endothelial cells, leading to a decrease in eNOS expression and NO secretion.
Studies have also shown that SUA has a natural antioxidant effect. 52 However, when SUA levels are too high, it may activate oxidative stress response, promoting the occurrence and development of cardiovascular disease.53,54 Xanthine oxidase (XO) is characterized by low specificity that can catalyze both hypoxanthine and xanthine to produce uric acid. 55 XO produces reactive oxygen species (ROS), which is an unstable strong oxidant and can oxidize cells and cause damage, such as superoxide anions or hydrogen peroxide in vascular endothelial cells with the participation of oxygen. 56 Therefore, high levels of SUA are usually accompanied by an increase in XO activity, resulting in the production of more ROS. 57 Some studies have shown uric acid can increase the expression and activity of NADPH oxidase to produce more ROS, stimulating an activation of p38 MAPK and ERK 1/2 pathway, thereby promoting the proliferation of VSMCs.58,59
This study has several strengths and limitations. Strengths include the use of publicly available GWAS data, multiple MR sensitivity analyses, and an exploratory NHANES analysis that was clearly separated from PAH diagnosis. Important limitations remain. First, the availability of suitable PAH GWAS datasets is limited; despite searching public GWAS resources, we did not identify a newer, large, fully compatible PAH GWAS for replacement or external replication. Second, GCST90018977 was a cross-population SUA GWAS and was therefore used only as exploratory secondary evidence. Third, the reverse MR analysis was removed because PAH yielded too few genome-wide significant instruments. Fourth, NHANES lacks right heart catheterization data and verified PAH diagnoses, so the NHANES analysis reflects a high-risk phenotype and provides only partial support for the MR findings. Fifth, PAH phenotype classification in GWAS remains challenging because PAH is rare and misclassification across pulmonary hypertension subtypes may bias genetic analyses. Such nondifferential outcome misclassification would generally be expected to attenuate associations toward the null, although the direction and magnitude of bias cannot be fully determined in summary-level GWAS data. Future datasets should use standardized right heart catheterization-confirmed phenotyping and detailed subtype classification. Finally, residual pleiotropy and population-specific effects cannot be fully excluded.
5. Conclusion
Our study supports a positive genetic association between higher SUA and increased PAH risk based on MR analysis. The NHANES analysis provides only exploratory population-level context in participants with a higher PAH-related risk-factor burden and does not validate or replicate the MR findings. Further studies are needed to confirm these findings in populations of different ancestry and to elucidate the underlying biological mechanisms.
Supplemental material
Supplemental material - Genetically predicted serum uric acid and pulmonary arterial hypertension: Mendelian randomization with an exploratory NHANES risk-burden analysis
Supplemental material for Genetically predicted serum uric acid and pulmonary arterial hypertension: Mendelian randomization with an exploratory NHANES risk-burden analysis by Huabin He, Zhekang Liu, Qingyun Yu, Qingan Fu, and Huangxin Zhu in Science Progress.
Supplemental material
Supplemental material - Genetically predicted serum uric acid and pulmonary arterial hypertension: Mendelian randomization with an exploratory NHANES risk-burden analysis
Supplemental material for Genetically predicted serum uric acid and pulmonary arterial hypertension: Mendelian randomization with an exploratory NHANES risk-burden analysis by Huabin He, Zhekang Liu, Qingyun Yu, Qingan Fu, and Huangxin Zhu in Science Progress.
Footnotes
Acknowledgments
We want to acknowledge the participants and investigators of all the GWAS used in this study.
Ethical considerations
No additional ethical approval was deemed necessary for this particular study.
Consent to participate
This study utilized data from participant studies that were publicly available and approved by an ethical standards committee for human experimentation.
Author contributions
HH and ZL: designed the study, performed the mendelian randomization analysis and NHANES analysis and drafted the first version of the manuscript. QY: draw figures. QF and HZ: designed the study, performed the mendelian randomization study and revised the manuscript. All authors have given approval to the final version of the paper.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The corresponding author is available to provide the data supporting the conclusions of this study upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.