
Abstract
Keratin from wool fibers was extracted with different extraction methods, for example oxidation, reduction, sulfitolysis, and superheated water hydrolysis.
Different samples of extracted keratin were characterized by molecular weight determination, FT-IR and NIR spectroscopy, amino acid analysis, and thermal behavior.
While using oxidation, reduction, and sulfitolysis, only the cleavage of disulfide bonds takes place; keratin hydrolysis leads to the breaking of peptide bonds with the formation of low molecular weight proteins and peptides. In the FT-IR spectra of keratoses, the formation of cysteic acid appears, as well as the formation of Bunte salts (–S–SO3–) after the cleavage of disulfide bonds by sulfitolysis. The amino acid composition confirms the transformation of amino acid cystine, which is totally converted into cysteic acid following oxidative extraction and almost completely destroyed during superheated water hydrolysis. Thermal behavior shows that keratoses, which are characterized by stronger ionic interaction and higher molecular weight, are the most temperature stable keratin, while hydrolyzed wool shows a poor thermal stability.
Keratins are a group of fibrous proteins produced in some epithelial cells of reptiles, birds, and mammals. These proteins are abundantly present in nature and constitute the major part of hair, wool, horns, nails, feathers, and the stratum corneum of the skin. Keratin is found in various sources in nature, equating to more than 5,000,000 ton/year and is available from textile industries, butchery, and breeding.1,2
These proteins are characterized by a high sulfur content and by the presence of strong disulfide bonds that make them water-insoluble and resistant to different chemical agents. There are different viewpoints to classifying keratins. Keratin has been classified as “hard keratin” and “soft keratin” according to its physiochemical properties, particularly on the basis of its sulfur content. It can also be classified as type I (acidic keratin) and type II (neutral–alkaline keratin) on the basis of the predominant amino acids and α-keratin and β-keratin on the basis of its secondary structure. Keratin can also be distinguished according to its biosynthesis and molecular weight.3–5
Keratin extracted from wool using different extraction processes can find applications in numerous sectors, such as biomedical, filtering, cosmetic, agricultural, and so on 6 , on the basis of its physiochemical properties. As a biobased material, wool keratin is biocompatible, biodegradable, and able to support cell attachment and spreading. 7 Indeed, wool keratins contain several peptide binding motifs, such as leucine-aspartic acid-valine (LDV) and glutamic acid-aspartic acid-serine (EDS), that support attachment of a wide variety of cell types. 8 Keratin is able to bind charged species such as heavy metal ions (copper, chromium, lead, mercury, etc.). It can also be used to remove formaldehyde and other volatile organic compounds (voc) from the environment. 9 Keratins have various applications in the cosmetic industry for the treatment of the skin and hair. 10 Keratin from waste greasy wool hydrolyzed with superheated water has been used for the preparation of organic fertilizers, with nitrogen release in the soil depending on the hydrolysis temperature and time.11,12
Large amounts of keratin are available from the coarse wool that is not suitable for spinning applications and wool waste from textile industries. Washed wool contains dominantly pure protein and it is formed of a histological structure that is made of three main morphological components: namely, the cuticle, the cell membrane complex, and the cortex. The cuticle of the wool fiber consists of a thin envelope of flat overlapping “cuticle cells” arranged like roof tiles surrounding the cortex, which is made up of spindle-shape “cortical cells” oriented parallel to the fiber axis. The cell membrane complex, sometimes referred to as intercellular cement, performs the function of cementing cortical and cuticle cells together.
Keratin can be obtained by different extraction methods. Keratin extraction may take place after cleavage of disulfide bonds and this can be achieved by oxidation, reduction, or sulfitolysis.
By oxidative extraction using peracetic or performic acids, which oxidize cystine to cysteic acid, keratoses are obtained. 13 Keratoses are water-soluble and cannot participate in cross-linking reactions with reformation of the disulfide bridges. For this reason, these biochemical materials can be degraded relatively fast in vivo compared to other extracted keratins, that is in the order of days to weeks. 14 Reductive wool extraction with an aqueous mixture of urea and thiols has been proposed by many authors.15,16 The reaction mechanism consists of two reactions of nucleophilic displacement, and the thiol groups obtained (cysteine residues) are rapidly oxidized by air. Sulfitolysis describes the cleavage of a disulfide by sulfite to give a thiol and a S− sulfonate anion (or Bunte salt). The sulfitolysis of cystine has been extensively studied by Cecil and his co-workers.17,18 While using the previous protein extraction methods the molecular weight of extracted proteins remains unchanged, wool protein hydrolysis leads to the breaking of peptide bonds with the formation of low molecular weight peptides. Wool hydrolysis can be achieved under basic or acidic conditions; complete acidic hydrolysis of wool keratin was used to study the chemical composition of wool fibers 19 while partial hydrolysis under controlled alkaline process conditions, with the aim of tailoring the extent of the peptide length, seems to focus the attention of many researchers.20,21 Green processes, solvent free, have been proposed for the hydrolysis of keratin. These treatments include hydrolysis with superheated water22,23 and enzymes. 24
In this paper, keratin extracted using different extraction methods (e.g. oxidation, reduction, sulfitolysis, and superheated water hydrolysis) was characterized and compared in order to suggest more suitable applications in correlation with the extraction method.
Experimental details
Materials and methods
Wool fiber, 22.00 µm mean diameter, in the form of top (fiber sliver obtained from raw wool by scouring, carding, and combing processes) was supplied from The Woolmark Co., Italy.
All chemicals were of analytical grade and purchased from Sigma-Aldrich, unless otherwise specified.
The fiber sample was cleaned by Soxhlet extraction with petroleum ether to remove fatty matter and surfactants, washed with Milli-Q water, conditioned at 20℃, 65% RH for 24 h, and cut into snippets around 2 mm length before further treatment.
Keratin extraction processes were carried out by oxidation, 25 reduction, 26 sulfitolysis, 27 and superheated water hydrolysis.23,28
Extraction methods
Oxidative extraction
For the oxidative extraction, 4 g of wool were put in an aqueous solution of 2% w/v peracetic acid (36–40 wt% peracetic acid solution in acetic acid) for 24 h at room temperature (fiber to liquid ratio 1 : 50), then the oxidized sample (pH 2.05) was washed with Milli-Q water, dried, treated in a Linitest apparatus with an aqueous solution of tris(hydroxymethyl)aminomethane (Tris) at 1 M for 2 h at 37℃ with vigorous shaking, and filtered through a 120 mesh sieve. The filtrate was adjusted to pH 7, dialyzed in the cellulose tube (molecular weight cut off 12–14 kDa) for two days against the circulated system of distilled water, then centrifuged (12,000 rpm, 15 min) to remove the precipitate.
Reductive extraction
For the reductive extraction, 4 g of wool were shaken in a 100 mL aqueous solution (fiber to liquid ratio 1 : 25) of urea (8 M), Tris (0.5 M), dithiothreitol (DTT, 0.14 M), and ethylenediaminetetraacetic acid (EDTA, 6 mM), adjusted to pH 8.6 with HCl, in a Linitest apparatus for 2.5 h at 25℃ and then filtered through a 120 mesh sieve. The filtrate was dialyzed in a cellulose tube (molecular weight cut off 12–14 kDa) for two days against the circulated system of distilled water and filtered (5 µm pore size) to remove insoluble parts.
Sulfitolysis
4 g of wool fibers were treated with 100 mL of aqueous solution containing urea (8 M), sodium metabisulfite (0.5 M), adjusted to pH 7 with NaOH (5 M) under shaking using a Linitest apparatus for 2.5 h at 65℃. The mixture was filtered through a 5 µm pore-size filter and dialyzed with the cellulose dialysis tube (molecular weight cut off 12–14 kDa) against distilled water in a circulating system for two days.
Superheated water hydrolysis
Keratin hydrolysate was prepared by treating 40 g of wool in 150 mL superheated water, for 30 min at 170℃, using a 400 mL ceramic autoclave placed in a microwave oven, Milestone Ethos 1600 (Milestone S.p.A., Bergamo, Italy). At the end of the process, the autoclave was cooled and the resulting product was filtered with a 120 mesh stainless steel sieve. The liquid phase was further filtered using a 0.65 µm pore size, tangential flow filter (Millipore Pellicon XL).
All keratins obtained with different extraction methods were freeze-dried for successive characterizations.
Characterization
Extraction yield
The extraction yield of the protein from the wool sample was evaluated using the following equation
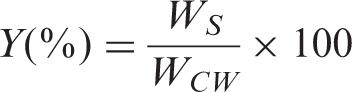
Molecular weight determination
The molecular weights of samples were determined by SDS-PAGE analysis. Keratin freeze-dried samples were extracted in a solution containing Tris–HCl (550 mM, pH 8.6), dithiothreitol (DTT, 140 mM), ethylenediaminetetraacetic acid (EDTA, 5 mM), and urea (8 M) under nitrogen atmosphere for 4 h. The SDS-PAGE was performed according to Laemmli’s method 29 using XcellLock Mini-Cell (Invitrogen) on 12% polyacrylamide gels.
Amino acid analysis
All the keratin-based material produced was hydrolyzed in HCl at 6 M for 24 h at 110℃ under nitrogen atmosphere. Free amino acid residues were derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC, Waters) and eluted on a reversed-phase column. An Alliance high performance liquid chromatograph (HPLC) (Waters) was used; amino acids were detected at 254 nm. The quantitative amino acid composition, expressed as mole% for each amino acid, was determined by external standard calibration using Amino Acid Standard H (Pierce), cysteic acid, and lanthionine.
Fourier transform infrared analysis (FT-IR)
Spectroscopic evaluation of extracted samples was carried out by FT-IR, using a Nexus Thermo Nicolet Spectrometer 510 P in attenuated total reflectance (ATR) mode using a diamond crystal. One hundred scans were taken from 4000 to 650 cm−1 with a resolution of 4 cm−1 and a gain of eight.
Near infrared (NIR) analysis
NIR spectra were acquired in the spectral region between 10,000 and 3700 cm−1, with an FT-NIR spectrometer (Perkin-Elmer) System, model Spectrum IdentiCheck (Monza, Italy), equipped with an IdentiCheck Reflectance Accessory (ICRA). For spectra acquisition, samples were placed on the 12 mm round window of the instrument; a mirror was used to reflect the beam back to the integrating sphere.
Differential scanning calorimetry (DSC)
DSC analysis was performed with a Mettler Toledo DSC 821 calorimeter calibrated by an indium standard. The calorimeter cells were flushed with 100 mL min−1 nitrogen. The runs were performed on the conditioned samples (20℃, 65% RH) from 30 to 400℃, at a heating rate of 5℃ min−1.
Thermogravimetric analysis (TGA)
TGA was performed with a Mettler Toledo TGA 850 analyzer. The temperature range was from 30 to 450℃ with a heating rate of 5℃ min−1 in a nitrogen atmosphere. About 3 mg of sample were used in each test using Al2O3 crucibles. The data were collected on a computer with the Mettler Toledo STARe System.
Results and discussion
Extraction yield
Extraction yield of different extracted samples

In general, there are not large differences in the extraction yield between different extraction methods.
The results are in agreement with data obtained by different authors. Because of the separation of oxidized wool into different fractions on the basis of solubility, the keratoses obtained are about 30% of the original wool; 19 yields ranging from 30–45% w/w were obtained by Yamauchi and Yamauchi 26 using reductive extraction of wool keratin, although other authors found bigger yields 30 using mercaptoethanol or thioglycolic acid 31 as reductive agents.
Similar yields (around 30% w/w) were obtained for sulfitolysis, 27 while the extraction yield using superheated water hydrolysis is related to temperature, reaction time, and material to liquor ratio. 23
Molecular weight (SDS-Page)
Figure 1 shows the electrophoresis pattern of all keratin extracted samples compared with the original wool as a reference. The wool sample (Lane 2) shows two important protein fractions at 67 and 43 kDa, which correspond to the low-sulfur (LS) proteins of the intermediate filaments present in the wool cortex and the protein fraction of the high-sulfur (HS) proteins in the cuticle, and different bands at a molecular weight lower than 10 kDa, corresponding to the high glycine, tyrosine protein of the matrix between cuticular and cortical cells.
32
Gel-electrophoresis pattern of the samples. Lane (1) molecular weight standard, (2) original wool, (3) keratoses, (4) kerateine, (5) sulfo-kerateine, (6) hydrolyzed keratin.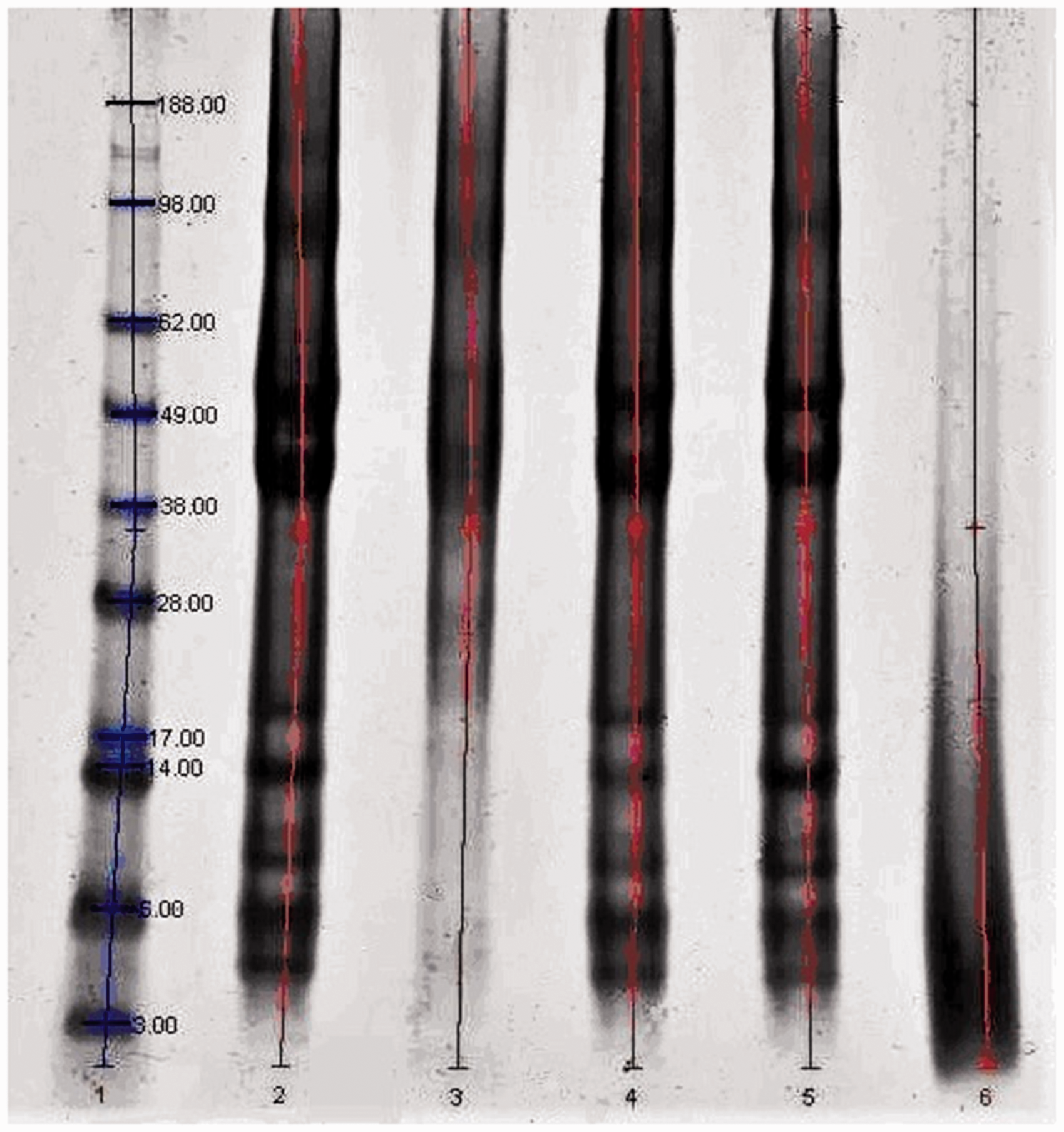
In the keratoses sample (Lane 3), the most important bands visible are at about 60 and 54 kDa, and they originate from the LS protein of the cortex, as already mentioned by other authors. 33 Due to the specific reaction of peracetic acid to the −S−S− bonds, in the electrophoresis pattern of keratoses, LS proteins appear as two bands well defined, so not damaged, and bands of other proteins (HS proteins) are not clearly visible.
The electrophoresis patterns of the superheated water hydrolyzed sample show that the strong bands of LS proteins disappeared and bands of high protein density appeared in the low fraction range at around 14–3 kDa. The loss of high molecular weight keratins indicates that the chemical structure of wool was significantly affected by the high temperature of the hydrolysis treatments, with the cleavage of peptide bonds. 32
Amino acid analysis
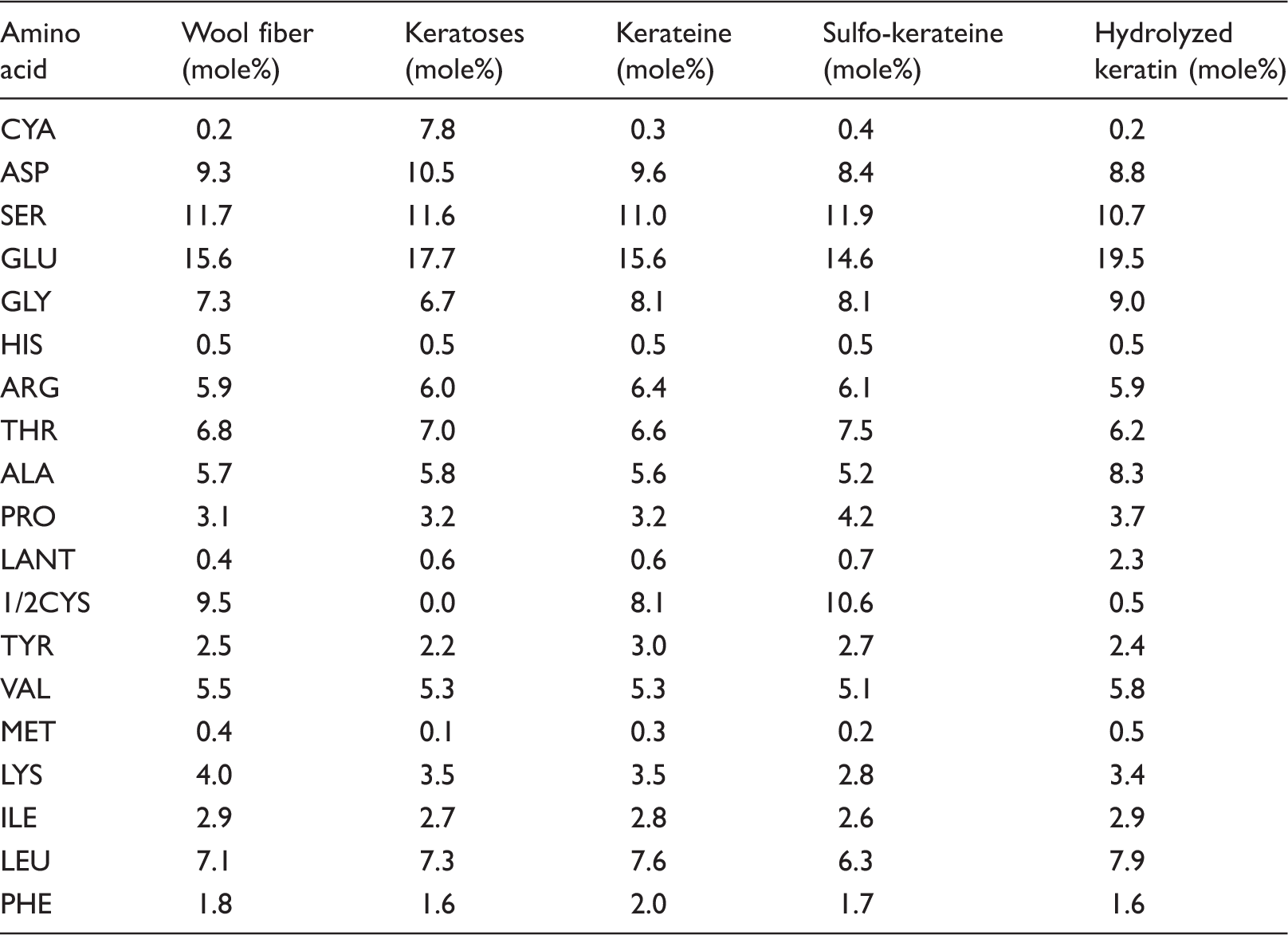
Amino acid analysis: keratoses, kerateine, sulfo-kerateine, and hydrolyzed keratin compared to amino acid analysis of original wool
During different extraction processes, cystine disulfide bonds are cleaved and new residues are formed according to the extraction process.
The chemical structures of amino acids that were affected by different extractions are shown in Figure 2.
The chemical structures of cystine, cysteine, cysteic acid, lanthionine, glycine, tyrosine, lysine, methionine. (a) Cystine, (b) Cysteine, (c) Cysteic acid (d) Lanthionine, (e) Glycine, (f) Tyrosine (g) Lysine and (h) Methionine.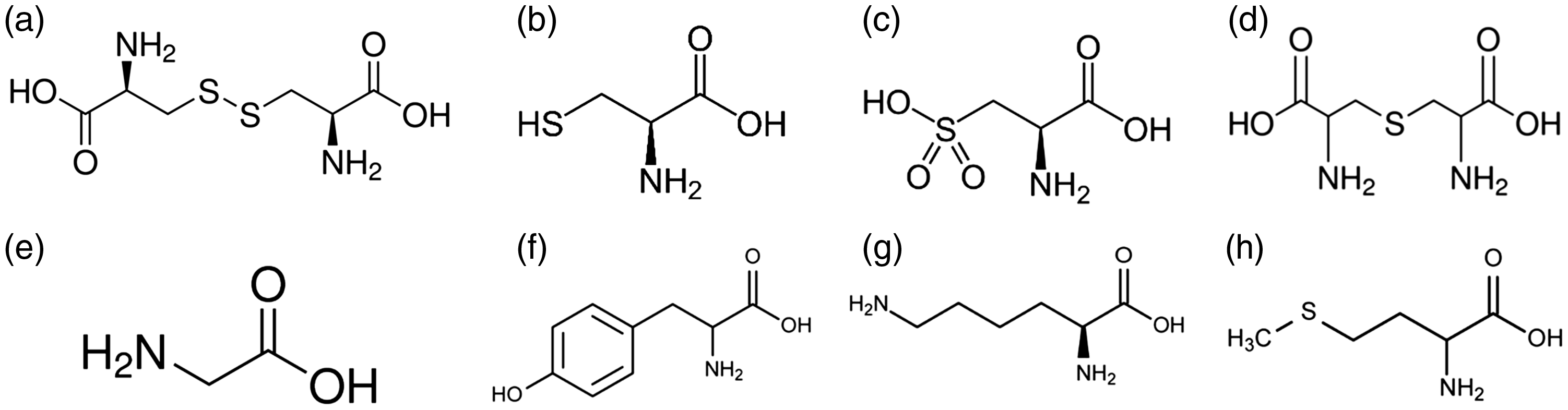
One of the main changes in amino acid composition among the samples is related to the conversion of cystine (and cysteine) to cysteic acid from the original wool to keratoses. Cysteic acid is found in very small amounts in the original wool where it is formed by the oxidizing effect of solar light on the fleece. Due to the oxidation process for the cleavage of disulfur bonds, the ½ cystine residues are completely converted to cysteic acid and there is no ½ cystine present in keratoses. In the amino acid composition of keratoses, a reduction in the amino acids having easily oxidizable sites is also observed 19 (such as tyrosine from 2.5 to 2.2 mole%, lysine from 4.0 to 3.5 mole%, and methionine from 0.4 to 0.1 mole%).
Moreover, glutamic acid, aspartic acid, leucine, and arginine, which are the amino acids contributing to the α-helix assembly of the LS proteins, were more abundant in keratoses than in the original wool, as confirmed by the molecular weight distribution where, especially, the LS proteins of the intermediate filament are present (see Figure 1). Only the lysine amount is not in agreement with this behavior. In keratoses, cysteic acid is present in a lower amount than ½ cystine in the original wool, probably due to the lower amount of cystine in LS proteins extracted by oxidation from cortical cells. 33 Moreover, glycine and tyrosine, mainly found in the low molecular weight proteins of the matrix between cuticular and cortical cells, are present in keratoses in a lower amount than in the original wool.
Other differences in amino acid composition are found between the original wool and hydrolyzed sample. In the hydrolyzed sample, the amount of ½ cystine was very low compared to the original wool. Most of the cystine was destroyed because of the temperature of the process, 23 and only a very small amount of cysteic acid, characteristic of undamaged wool, was found. The loss of cystine is associated with thermal treatment and the cleavage of –S–S– bonds results in the formation of lanthionine (2.3 mole%) through the formation of dehydroalanine and free sulfidryl groups. Dehydroalanine and the free sulfidryl group of cysteine can further react to form the irreversible cross-link –S– of lanthionine. H2S is also produced from the degradation of cystine. 19 In addition, small changes detected in the amount of the other amino acids in the hydrolyzed sample can be correlated with their thermal stability and secondarily with their solubility in water. Moreover, the breaking of disulfide bonds as a result of cystine destruction combined with the breaking of peptide bonds shown by electrophoresis (see Figure 1) caused a considerable amount of peptides and free amino acids to be dissolved in water.
No significant changes were found between the amino acid composition of kerateine and sulfo- kerateine compared with the original wool.
FT-IR spectroscopy
Figure 3 shows the infrared spectra of all extracted samples compared with the original wool spectrum.
Fourier transform infrared spectra of (a) wool, (b) keratoses, (c) kerateine, (d) sulfo-kerateine, (e) hydrolyzed keratin.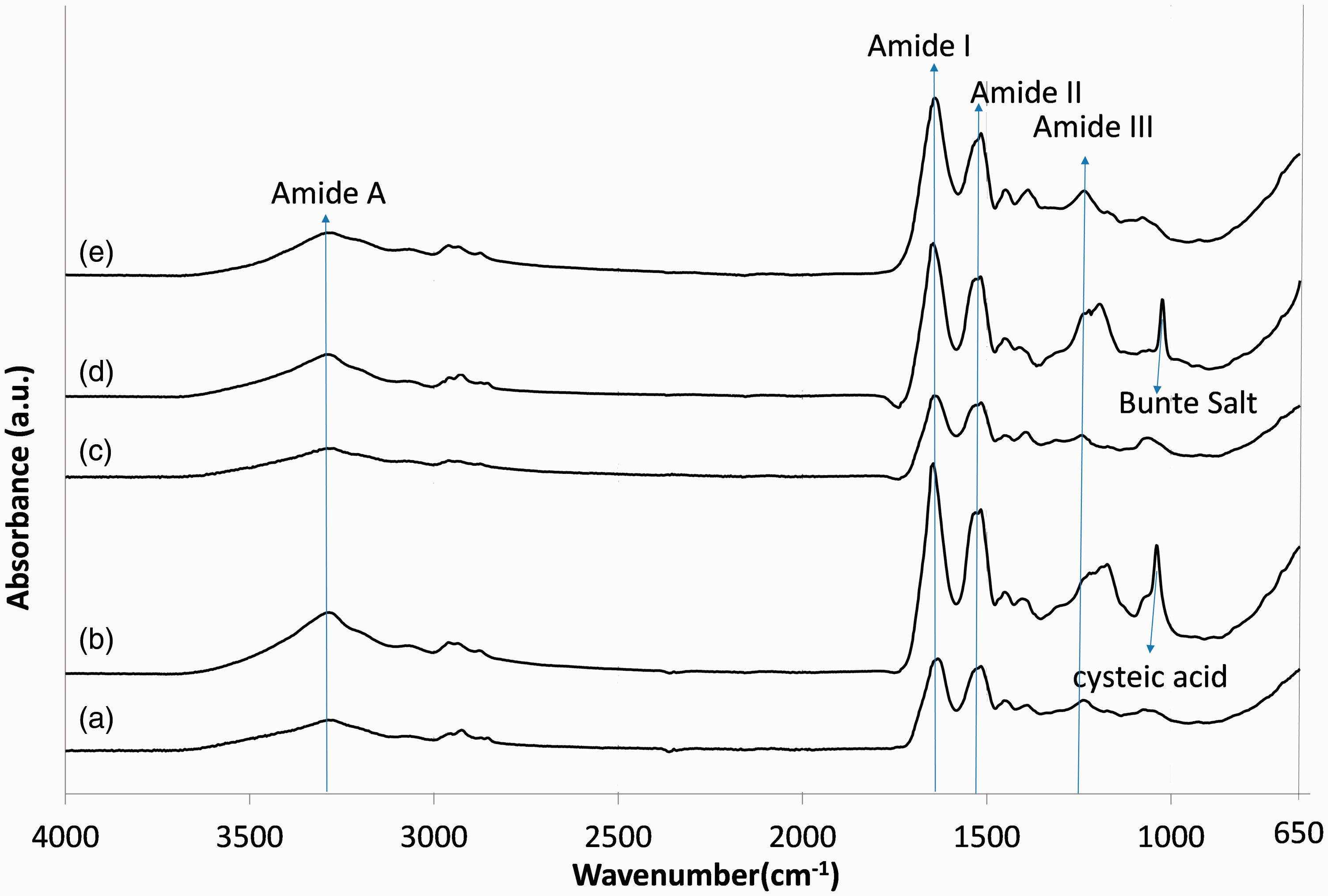
The infrared absorption spectra of all samples show characteristic absorption bands attributed to the peptide bonds (−CONH−). The vibration in the peptide bonds originate bands known as amide A, amide I, amide II, and amide III. The amide A band at 3282 cm−1 is connected with the stretching vibration of the N−H bond. Amide I, which is related mainly to the (C=O) stretching vibration, has an intense peak at 1630 cm−1. Amide II, which is related to the N−H band, has a peak at 1515 cm−1, while the amide III band at 1230 cm−1 is referred to as the in-phase combination of C−N stretching and N−H in-plane bending, with some attributed to the C−C stretching and C=O bending vibrations. 34
Peaks that appear between 1200 and 1000 cm−1 are attributed to the S−O vibration (see Figure 3). In particular, these peaks are present in the spectra of keratoses and sulfo-kerateine in comparison with the original wool. The absorption bands at 1174 and 1040 cm−1 in the keratoses spectra are related to the asymmetrical and symmetrical S−O vibrations of sulfonate (SO3−) in cysteic acid. 35 Because of the complete oxidation of cystine to cysteic acid during the oxidation process to obtain keratoses, these two vibration bands appeared in the keratoses sample; while the absorption bands of cystine monoxide (–S–SO–) and cystine dioxide (–S–SO2–) at 1078 and 1120 cm−1, respectively, which are intermediate products in oxidation of cystine to cysteic acid, 19 are nearly not present in keratoses.
In the sulfo-kerateine spectrum, two absorption bands are clearly visible at 1022 and 1095 cm−1, assigned to the sulfur–oxygen vibration of sulfonate in the cysteine sulfunic acid (–S–SO3− Bunte salts).
In the spectra of kerateine and hydrolyzed keratin, no evident differences were found in comparison with the original wool spectrum.
NIR spectroscopy
Figure 4 shows the FT-NIR spectra of extracted samples compared with the original wool spectrum.
Near infrared spectra of (a) wool, (b) keratoses, (c) kerateine, (d) sulfo-kerateine, (e) hydrolyzed keratin.
The absorption bands in the near infrared region (between 10,000 and 3700 cm−1) are the result of overtones and combinations originating in the fundamental mid-range infrared region of the spectrum. The bonds involved are generally C–H, N–H, O–H, and so on. Before NIR analysis, all the samples were previously stored for 24 h in a standard atmosphere (20℃, 65% RH) in order to normalize the water amount.
The shoulder at 7000 cm−1 is related to the first overtone of the O–H stretching vibration of water and the band at 5200 cm−1 is assigned to a combination of the O–H stretch and H–O–H bending vibration of the hydroxyl group from water. The doublet at 5800 cm−1 is an overtone of the C–H stretch of the protein side chains and lipids. The bands between 5000 and 4000 cm−1 are attributed to the characteristic molecular conformation and amino acid composition of the keratin samples. In detail, the bands between 5000 and 4500 cm−1 are related to the secondary structure of the proteins, and the bands from 4500 to 4000 cm−1 are mainly attributed to the nature of the protein side chains. 2
By comparing the spectral absorption of all samples, as can be seen, there are small differences in the achieved spectra, which are related to the hydration state of the samples (shoulder at 7000 cm−1 and bond at 5200 cm−1). 36
Moreover, the second derivate spectra of wool and extracted keratin were compared (see Figure 5). The band at 4525 cm−1, which seems to be associated with the β-sheet structure, is similar and very low in all samples. Significant differences are visible in the region between 4500 to 4000 cm −1 that is considered to reflect the amino acid composition of samples.
37
Second derivative near infrared spectra of (a) wool, (b) keratoses, (c) kerateine, (d) sulfo-kerateine, (e) hydrolyzed keratin.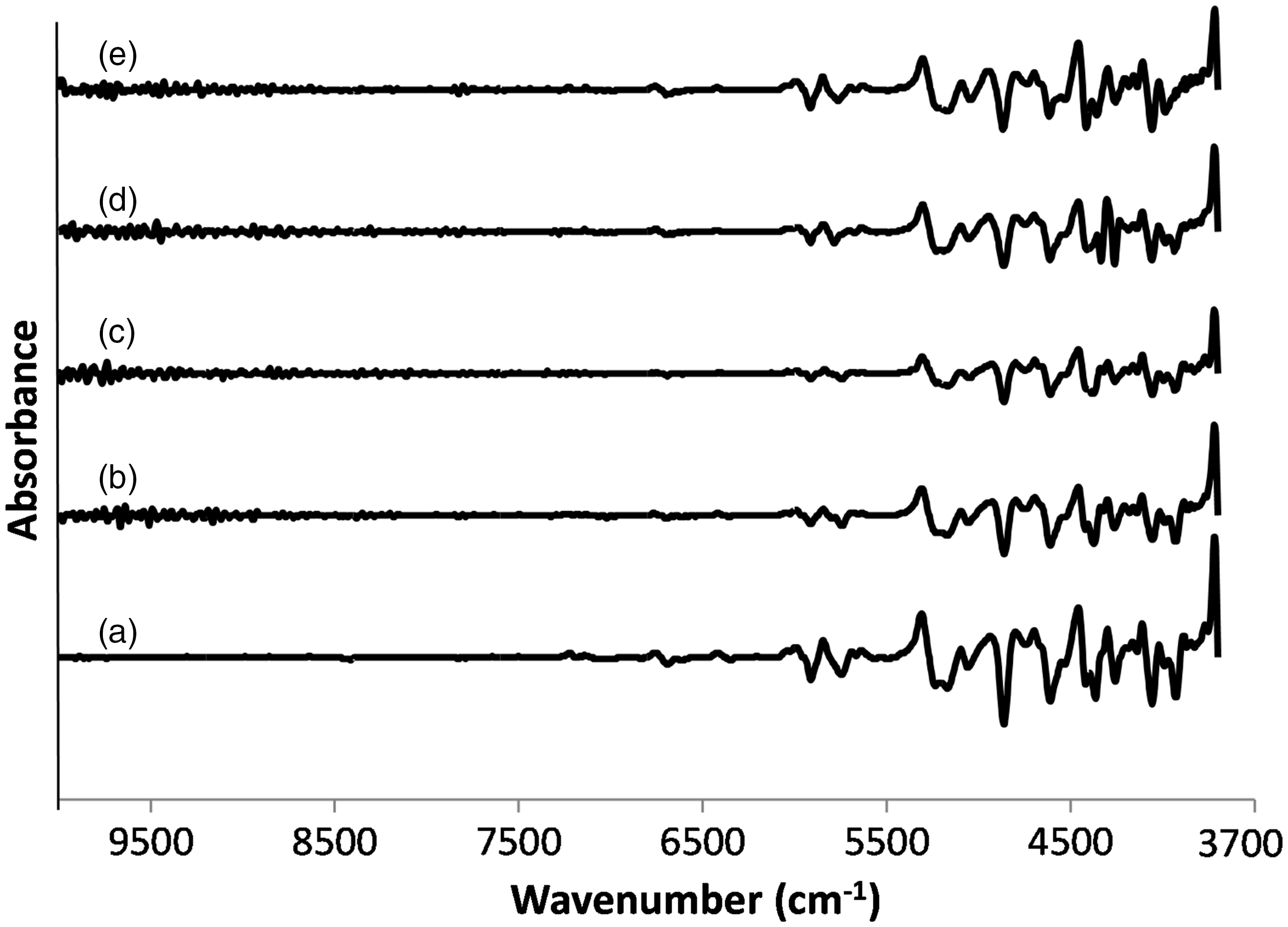
Thermal behavior
In Figure 6, the DSC thermograms of wool extracted or hydrolyzed in different ways are shown and compared with the original wool. In each thermogram, the first endotherm peak below 100℃ due to water evaporation is followed by peaks related to protein denaturation.
Differential scanning calorimetry curves of (a) wool, (b) keratoses, (c) kerateine, (d) sulfo-kerateine, (e) hydrolyzed keratin.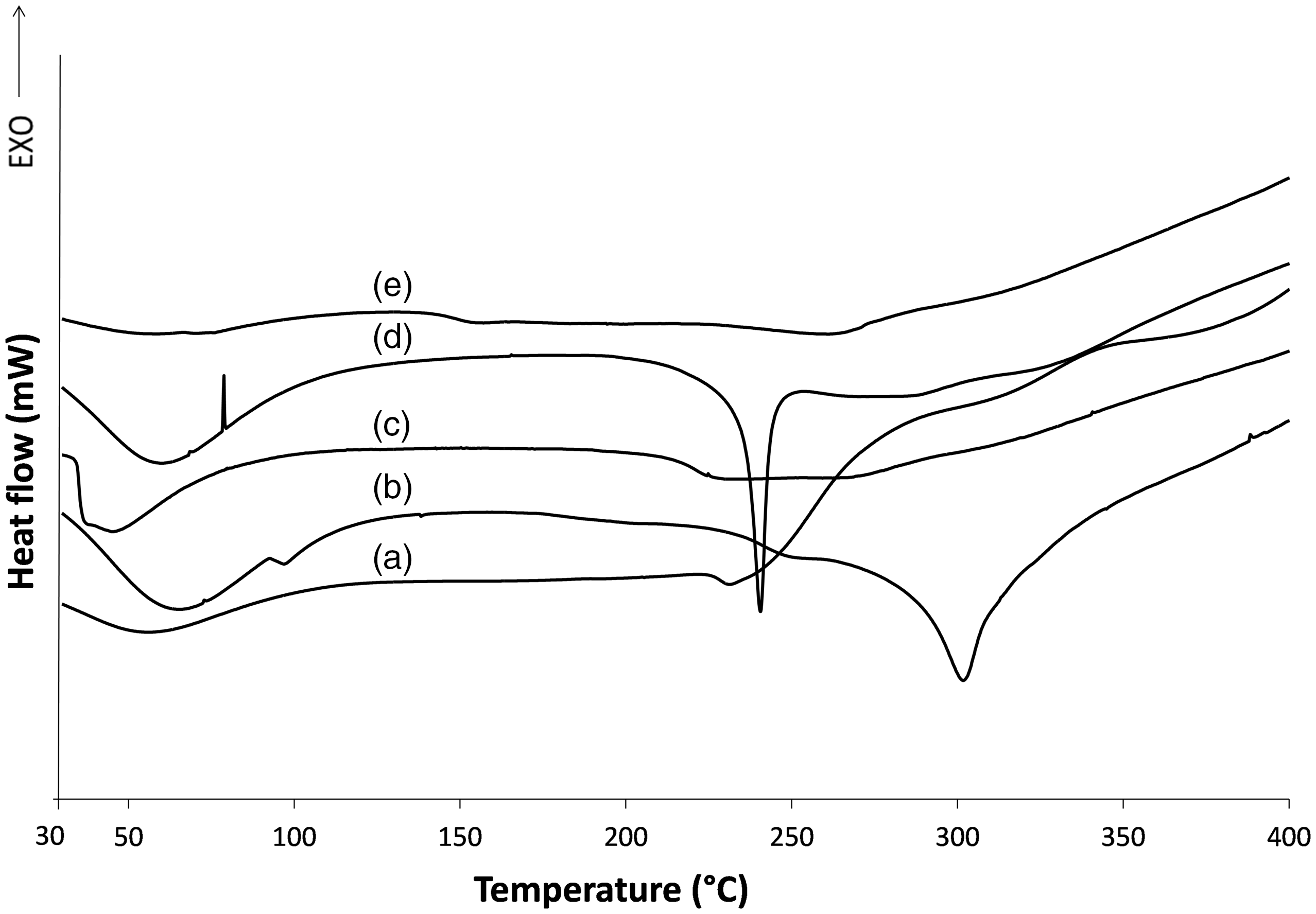
In the original wool, the α-helix denaturation of cortical cells takes place at about 230℃ with a peak that includes the denaturation of α-helical crystalline material in the domain of both ortho and para cortical cells. 38
The analytical traces of extracted keratin are very different, both for the temperature and for the form of denaturation peaks. A thermogram of the hydrolyzed wool does not show typical denaturation or degradation temperatures, such as keratoses or sulfo-kerateine traces, but broad peaks in a broad range at lower temperatures, indicating that the thermal stability of the hydrolyzed keratin is reduced with respect to keratoses or sulfo-kerateine. Similar results were obtained by Fortunati et al. comparing enzyme hydrolyzed keratin with sulfo-kerateine extracted from merino wool and brown alpaca fibers. 39 Also, kerateine, characterized by the re-oxidation of disulfur bonds from thiols, shows peaks that are not well defined in a broad temperature range.
The hydrogen and ionic bonds in sulfo-kerateine and keratoses proteins make them degrade at precise temperatures with well defined melting peaks, at about 240℃ for sulfo-kerateine and more than 300℃ for keratoses.
The higher thermal stability of keratoses and the lower thermal stability of hydrolyzed wool are confirmed by thermogravimetric analysis (see Figure 7).
Thermogravimetric analysis of extracted keratins.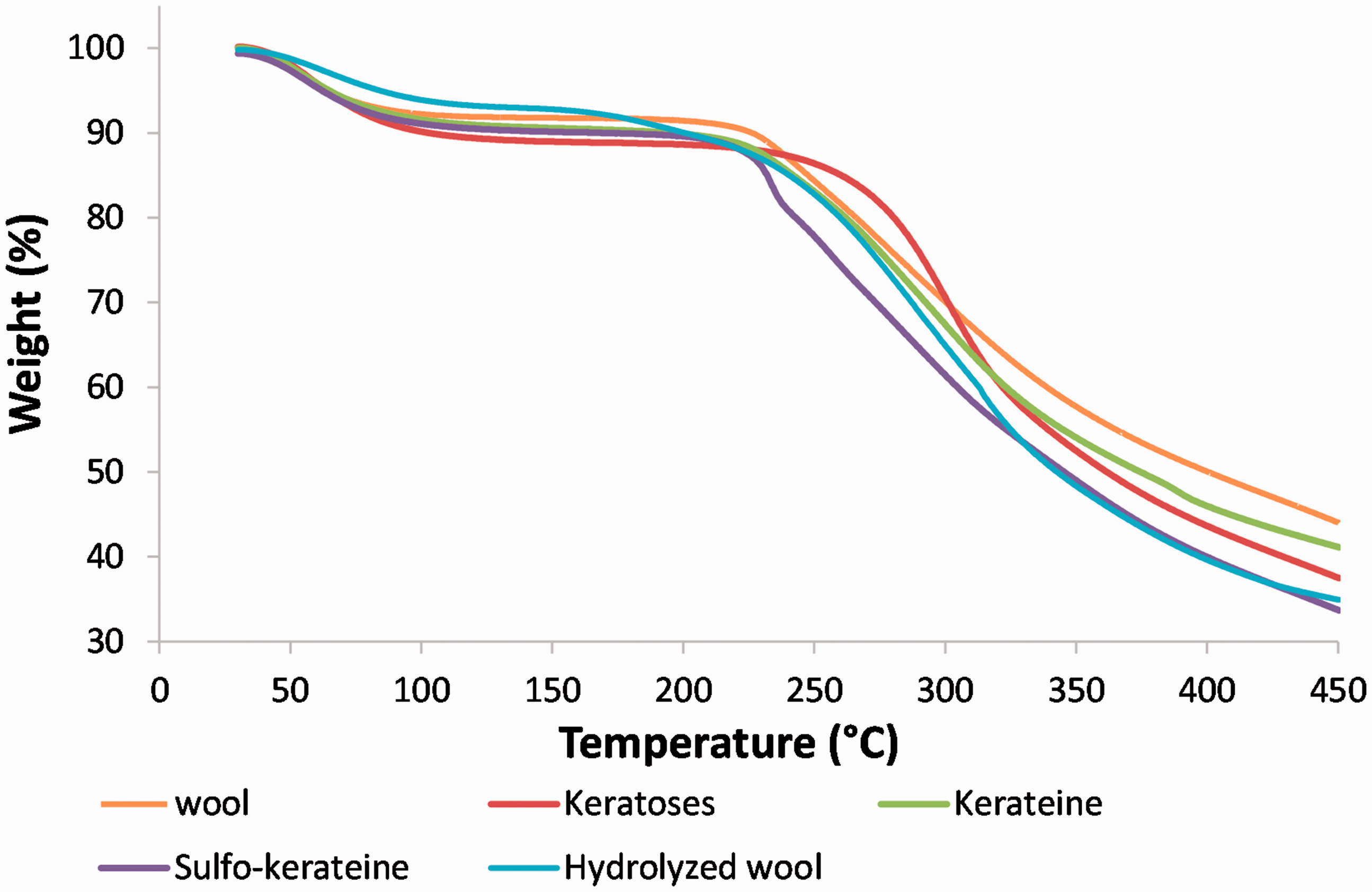
Conclusion
Keratin from wool fibers was extracted by oxidation, reduction, sulfitolysis, and superheated water hydrolysis, and the different extracts obtained were compared. Although extraction yields are quite similar, evident differences can be noticed in the time, cost, and environmental sustainability of the extraction methods. Among the selected extraction agents, sodium metabisulfite, used for sulfitolysis, seems to be the cheapest and least harmful agent in comparison with peracetic acid and dithiothreitol, utilized for oxidative and reductive extraction, respectively, being similar to the molecular weights of the products obtained. Superheated water hydrolysis highly decreases the molecular weight of the peptides obtained, with loss of the most temperature sensitive amino acids, but it is a cheap and environmentally friendly method, easy to implement on a large scale. 11
Although extracted keratins via oxidation, reduction, and sulfitolysis can be used in highly technological fields, such as the biomedical sector for scaffold production or the filtration sector, where the mechanical characteristics of the proposed materials are also to be tailored, the hydrolysis of keratin represents one of the most promising ways to extend the practical application of keratin in the agricultural, animal feed, or cosmetic fields. Other characteristics, such as solubility, the presence of polar groups (e.g. cysteic acid in keratoses), reformation of the disulfur bond capacity, can be considered to use keratin itself extracted in different ways or extracted keratin in a blend with other polymers, for tailoring specific material characteristics.
Footnotes
Acknowledgements
Authors would like to thank the Sustainable Management and Design for Textiles (SMDTex) Erasmus Mundus Joint for doctorate program of Hossein Rajabinejad.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.